OBJETIVO
Realizar un ejercicio de análisis complejo de datos obtenidos a partir de experimentos de espectroscopía infrarroja realizados en el laboratorio.
ELEMENTOS DE PARTIDA
Disponemos de dos espectros de infrarrojo de la proteína SmelDhp (una dihidropirimidinasa de Sinorhizobium meliloti) en ausencia y presencia de 1 mM Zn2+ a pH 7.0 y temperatura 20 ºC. Fichero EXCEL de ayuda para generar el fichero PKS de parámetros iniciales del ajuste de bandas componentes de la banda amida I.
EVALUACIÓN
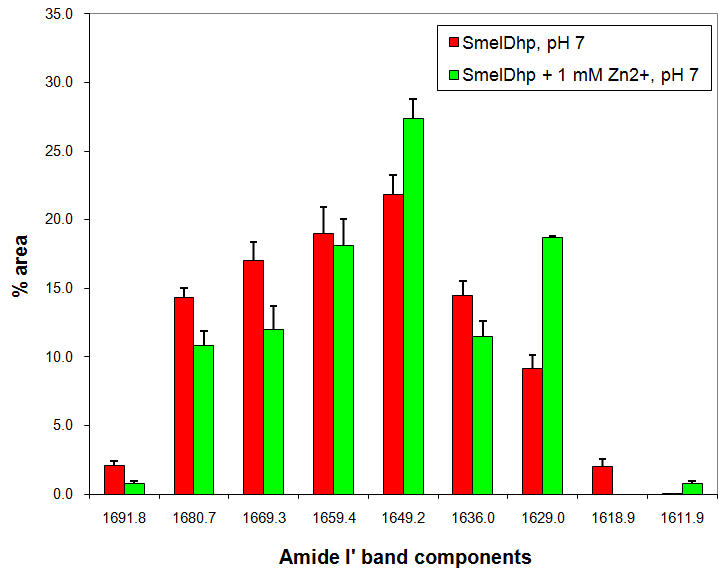
Se pedirá a cada alumno que muestre en un gráfico de barras los resultados del análisis de estimación de estructura secundaria de la banda AMIDA I' de ambos espectros usando métodos de mínimos cuadrados, como se explica en el tutorial que se muestra a continuación.
TUTORIAL PARA LLEVAR A CABO UNA ESTIMACIÓN DE ESTRUCTURA EN BASE A DATOS DE FTIR
La mayoría de los programas que debemos utilizar han de ejecutarse en un emulador de MS-DOS, que ya hemos usado anteriormente. Empezaremos descargando y descomprimiento en el disco duro C:\ de nuestro computador de trabajo (S.O. Windows XP, Windows Server 2003 o 2008, Vista o 7 a 32 o a 64 bits) el emulador que permite ejecutar los programas: Gplotc, Ramopn, xformatn, Spectracalc.
1. Transformación de ficheros a distintos formatos.
El espectrómetro de FT-IR genera ficheros de registro de los datos con extensión *.spc. Lo primero que haremos es generar otros ficheros con los mismos datos que hemos adquirido, pero con un formato que entiendan otros programas que usaremos para llevar a cabo las estimaciones de estructura secundaria. Para ello utilizaremos los programas del tutorial hasta generar ficheros *.dt.
2. Reconstrucción de los espectros empleando algoritmos de máxima entropía.
Antes de proceder al ajuste de bandas componentes de la banda Amida I' se llevará a cabo un procedimiento para eliminar ruido del espectro original, denominado reconstrucción del espectro empleando algoritmos de máxima entropía. Este proceso se lleva a cabo en el programa SpectraCalc, que debes ejecutar desde emulador DOSBox como se muestra en la siguiente figura:
La primera pantalla que muestra el programa Spectra Calc es la siguiente ...
Pulsando F2 desplegamos el Menu:
En "Environment" seleccionamos el "Directory" en el que estamos trabajando o en el que queramos trabajar,
por ejemplo c:\sc\data

A continuación definiremos los límites, en números de onda, del espectro que queremos reconstruir, por ejemplo entre 1700 y 1600 cm-1, ya que nos interesa estudiar la banda amida I, que se localiza entre 1700 y 1600 cm-1.
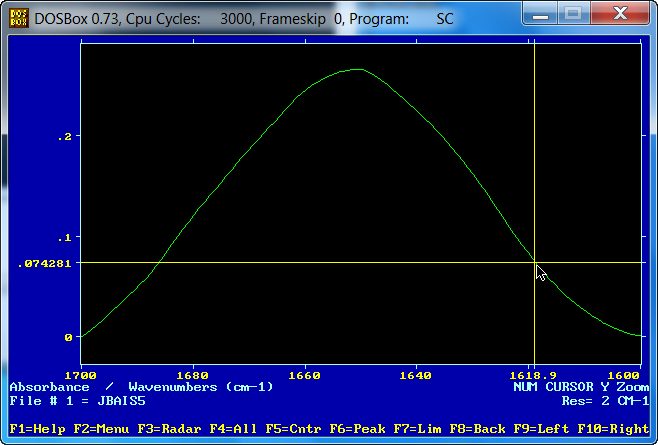
Cargaremos con SpectraCal el fichero que queremos reconstruir, por ejemplo: JBAIS5.SPC como se muestra en la siguiente figura:


En el menu de Aritmetic seleccionamos DoProgram ...
De este menú elegimos MSMOOTH ...

... y de la siguiente pantalla elegimos "Nsmooth"
Elegimos como forma de banda Gaussiana ...
... y además elegimos escribir la anchura de la banda a recosntruir en 12 cm-1.


El espectro resultante del proceso de reconstrucción aparece en color naranja en la siguiente figura ...
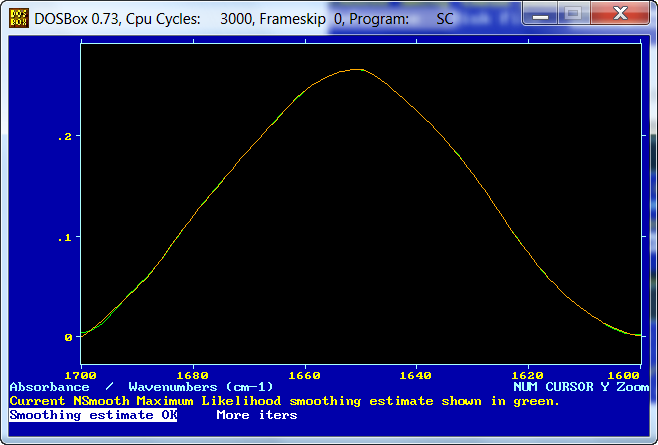
La siguiente pantalla nos proporciona información sobre la relación ruido/señal resultante "RMS= .00073682"
En el Menú de File, presionamos Save, para guardar el nuevo espectro, sobre el que llevaremos a cabo el proceso de ajuste de bandas componentes a la banda Amida I'. A continuación repetiremos este mismo proceso con el espectro JBAPS1.spc
3. Cálculo de los espectros derivada y desconvolución utilizando los programas Ramopn y Gplotc.
La estimación de estructura secundaria por ajuste de bandas componentes de la banda Amida I' se realizará utilizando los espectros que acabamos de generar tras el proceso de reconstrucción empleando algoritmos de máxima entropia, o sea, sobre los ficheros jbaiSM5.spc (SmelDhp, pH 7) y jbapSM1.spc (SmelDhp + 1 mM Zn2+, pH 7). Nótese que los ficheros resultantes del "alisado" contienen una M en su nombre.
Para calcular los espectros DERIVADA y DESCONVOLUCIÓN utilizaremos el programa Ramopn que sólo lee dicheros *.dt, por lo que tendremos que transformar los ficheros jbaiSM5.spc (SmelDhp, pH 7) y jbapSM1.spc (SmelDhp + 1 mM Zn2+, pH 7) en *.dt como hicimos en la práctica 6A.
La primera pantalla que presenta el programa Ramopn es:
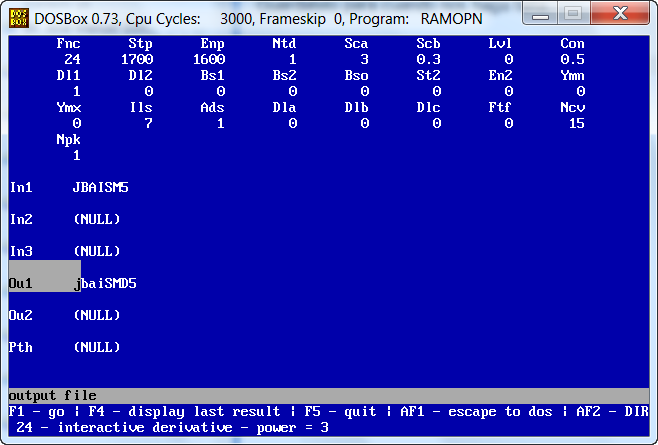
Obsérvese que la función 24 permite calcular la derivada. Los parámetros a incluir son Sca=3, Scb=0.3 y los extremos del espectro entre 1700 y 1600. Fijarse bien en que los números de cada campo coincidan con los que se observan en la pantalla. Presionando la tecla F1 calcularemos el espectro derivada.
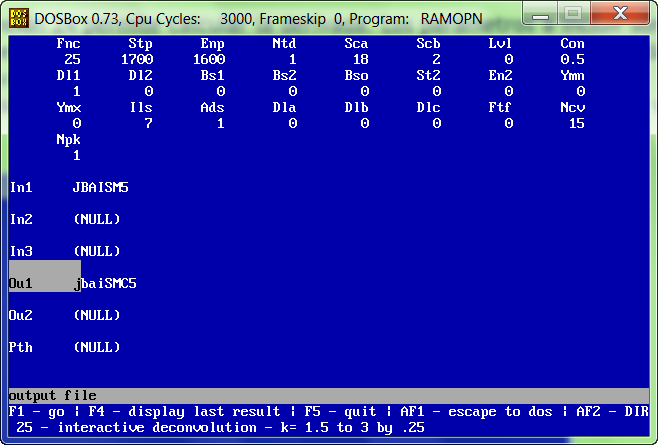
Para calcular la desconvolución utilizamos la función 25, con los parámetros que se ven en la siguiente pantalla:
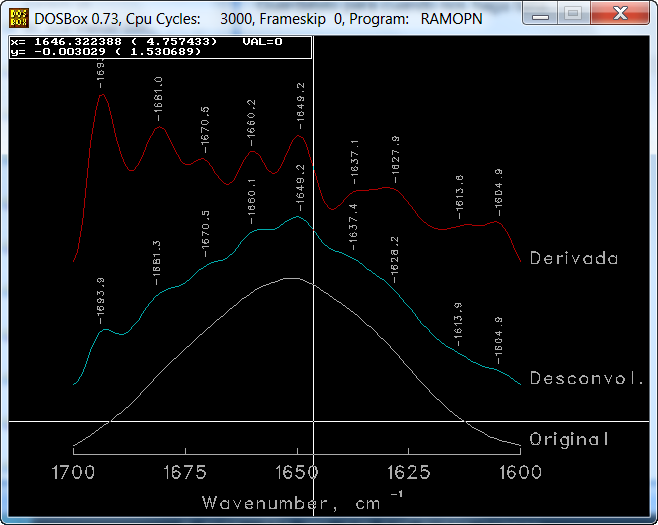
En el programa Gplotc, ubicado en el directorio c:\nrc podremos ver la posición del máximo (Alt+F8) de las bandas componentes de la banda Amida I' y el número de bandas componentes, entre otros parámetros útiles para construir el fichero *.pks a partir del cual hacer los ajustes.
4. Estimación de estructura secundaria de una proteína por ajuste de bandas componentes de la banda Amida I' de su espectro de FTIR.
Las soluciones matemáticas al ajuste de bandas componentes de una banda compleja serían infinitas a menos que nosotros impusiéramos restricciones en los parámetros de partida. Siempre se debe tener presente que los resultados matemáticos no siempre tienen sentido en términos biológicos. Debe quedar claro que estamos ante una metodología que permite ESTIMAR los porcentajes de estructura secundaria en proteínas solubles o de membrana. Nunca pensaremos en términos absolutos y asumiremos que es útil para detectar cambios relativos en los componentes de estructura secundaria como consecuencia de modificaciones del ambiente (pH, fuerza iónica, unión de un ligando, etc.).
Los parámetros iniciales que van a determinar las restricciones en el ajuste de bandas componentes de la banda amida I (1700-1600 cm-1) son:
- Número de bandas componentes, dicho parámetro vendrá determinado por los datos que nos proporcione la derivada y la desconvolución del espectro que hemos obtenido. En este ejemplo hemos usao en la derivada unos parámetros de 3 y 0.3 (función 25 del programa Ramopn) y en la desconvolución usaremos como parámetros 18 y 2 (función 24 del programa Ramopn).
- Anchura de la banda, en la bibliografía se recomienda que se calcule la anchura a mitad de altura de cada componente resultante en la derivada y se multiplique por 2,7. Sin embargo, no siempre esto es posible ya que en los componentes que detecta la derivada no siempre es posible realizar este cálculo. Queda a la elección del investigador la elección de dichas anchuras, cuando no es posible usar el criterio anterior.
- Altura de los componentes, para los componentes de los extremos y el mayor visto en desconvolución tomaremos el 90 % de la absorbancia del espectro original realizando una línea base entre 1600 y 1700 cm-1, y para los restantes componentes el 70 %.
- Forma de la banda, Mezcla Lorentziana/Gaussiana.
Siguiendo las indicaciones antes mencionadas generamos en cualquier editor de texto un fichero de extensión *.pks con la información similar a la que se presenta en esta figura pero obtenida para nuestros espectros problema:
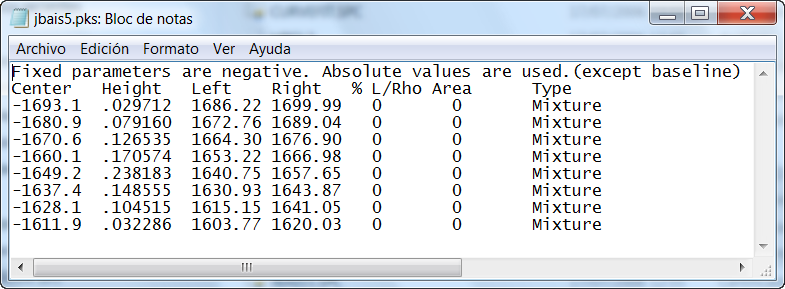
Este fichero *.pks será usado por SpectraCalc para llevar a cabo un ajuste de bandas componentes de la Amida I' como fichero de estimados iniciales.
Para realizar el ajuste empezaremos por cargar el fichero jbaiSM5.spc en SpectraCalc. Nos aparece la región del espectro comprendida entre 1700 y 1600 cm-1.
Ahora tomaremos una línea de base y después ajustamos la absorbancia de los extremos con
valor de cero.
Para conseguir la línea de base desplegamos el Menú:
"ARITMETIC"
"OTHER"
"BASELINE"
"TWO-POINT BASELINE CORRECTION"
Presionando en el extremo de 1700 la tecla F9 y en el 1600 la F10 y después "Enter" se consigue la línea base ...
Utilizando el fichero jbais5.pks, que contiene los parámetros iniciales,
procederemos a realizar el ajuste. De nuevo desplegamos el Menu:
"ARITMETIC"
"OTHER"
"BASELINE"
"CURVEFIT"
En una pantalla intermedia nos pregunta sobre el número de bandas
componentes con las que queremos realizar el ajuste y propone introducir "0" si
disponemos de un fichero *.pks con los parámetros iniciales que es nuestro caso.
El resultado obtenido se puede seguir en la siguiente pantalla ...
La pantalla siguiente nos pregunta el número de iteraciones que realizará en programa: 200
El espectro resultante después del primer ciclo de interaciones
(color blanco) queda superpuesto al espectro de partida (color verde). Se puede observar
que aun no existe una coincidencia total entre ambos espectros y por tanto aun es preciso
un segundo ciclo de iteraciones liberando en esta ocasión la posición de los
componentes.
Inicialmente debemos guardar el fichero de datos con los resultados del ajuste tras las
primeras 200 iteraciones. Para ello desplegamos los menús:
PARAMETERS
FILE PARAMS
jbaiS51.pks (nombre de fichero)
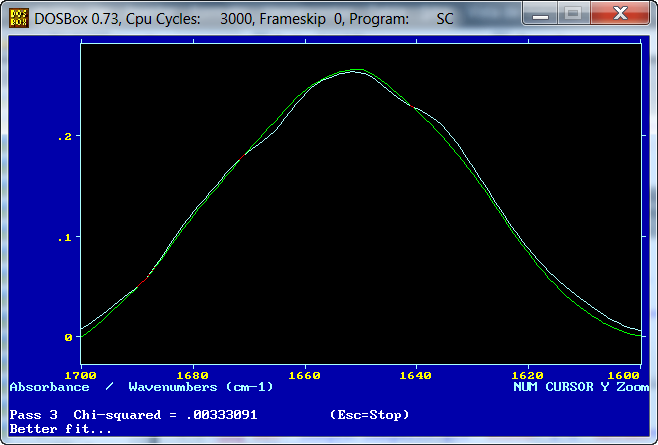
Finalizamos esta parte del protocolo pulsando "END"
Editamos el fichero jbaiS51.pks y liberamos la posición de componente para después
seguir otro proceso de 50 iteraciones.
Ahora realizamos el mismo proceso que cuando hemos llevado a cabo las primeras 200
iteraciones. O sea ...
ARITMETIC
OTHER
CURVEFIT
ENTER
0
jbaiS51.PKS
50 (ITERACIONES)
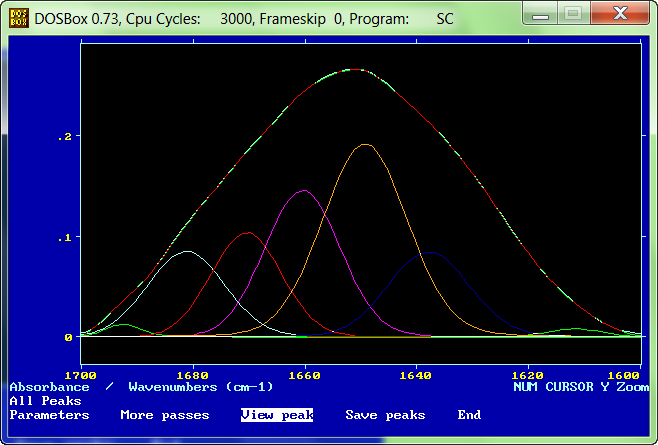
En el menú "View peaks" podemos ver los resulatdos del ajuste ...
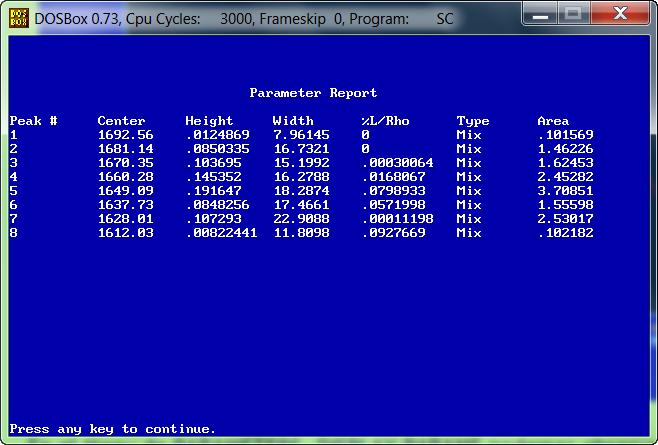
En el menú "Parameters" se ven los datos numéricos del ajuste ... Donde encontramos el área asignado a cada banda componente después de finalizado el ajuste.
Después de llevar a cabo el proceso de ajuste para ambos espectrsos, se muestra (a modo de ejemplo) una gráfica que presenta los resultados de estimación de motivos de estructura secundaria de la proteína SmelDhp en ausencia y presencia de 1 mM Zn2+.