Chuletas para computar... cuando la memoria falla!
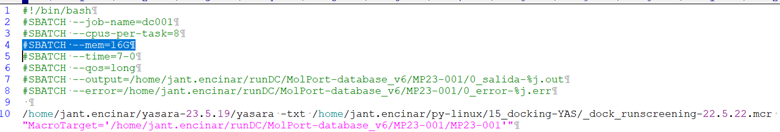
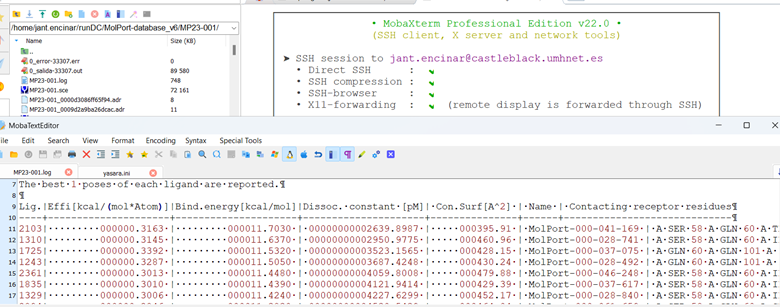
RUNNING YASARA's docking.
Cambiar el ancho del panel lateral derecho (internal GUI) de PyMol compilado y libre: set internal_gui_width, 300
Running YASARA on computer clusters
En el yasara.ini ===> Memory 10000

y 8 CPUs,

y el sbatch con 8 CPUs y 16G de memoria
Comando de PyMol que selecciona los aminoácidos a la distancia elegida a un aminoácido u objeto
# selecciona los residuos a una distancia inferior a 17 A del centro del obj01
select selcenter, (byres (obj01 around 17))
# selecciona los residuos a una distancia inferior a 8 A del centro del residuo Cys 215
select selcenter, (byres (br. (resi 215 and resn CYS) around 8))
show sticks, selcenter
# Stop the simulation if the distance between Lys 90 in Mol B and the ligand ATP in Mol C becomes >10 A:
dis = GroupDistance Res ATP Mol C, Res Lys 90 Mol B
if dis>10
break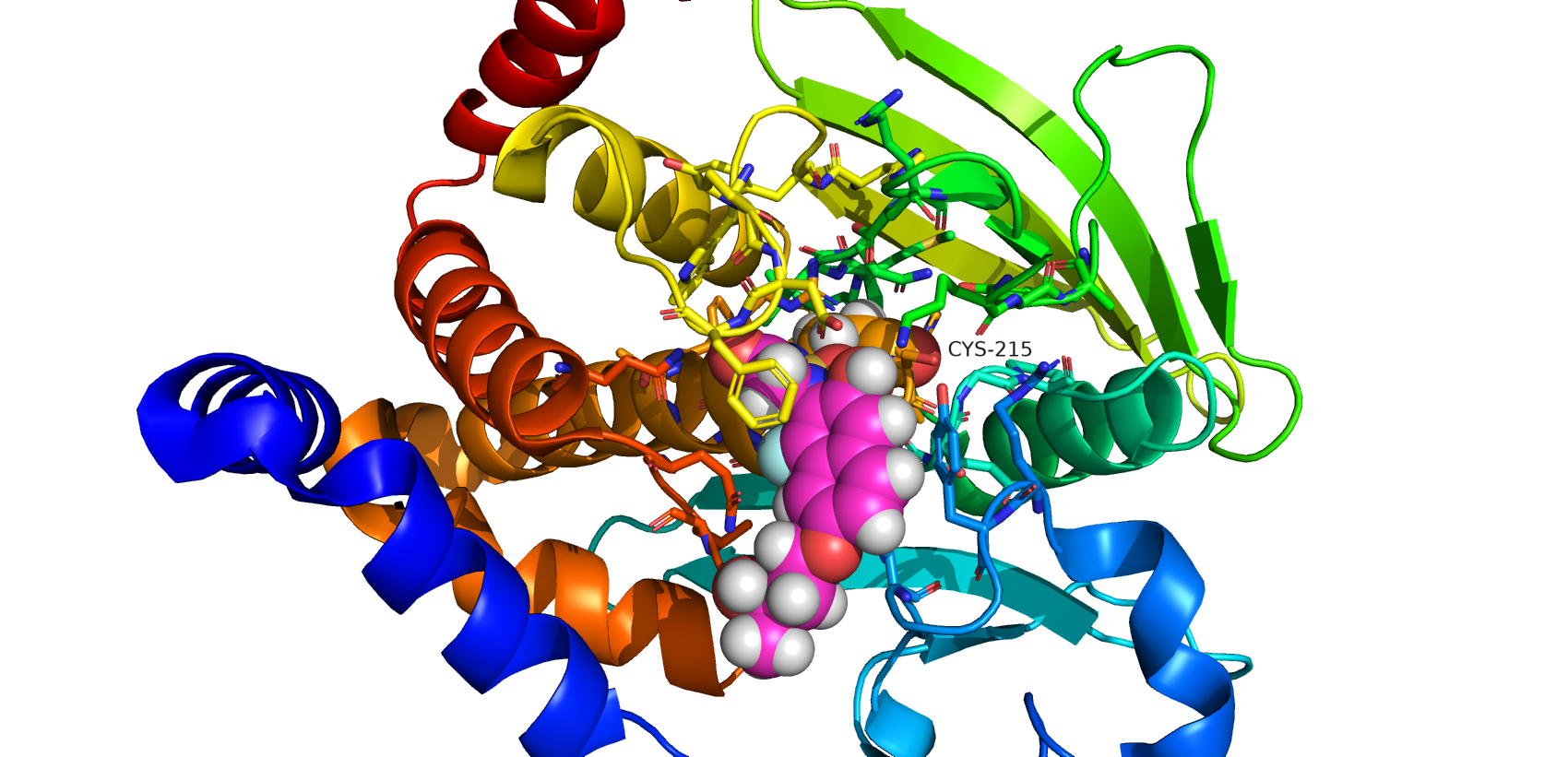
Sección de una proteína en PyMol en color gris
fetch 4ZYO, async=0
set ray_opaque_background, 0
color_h
as surface,
clip near , -50
set ray_interior_color, gray
set ray_shadows,0
ray
png D:\encinar\tempjant\02-images\4ZYO-structure.png, width=1200, height=1200, dpi=600, ray=1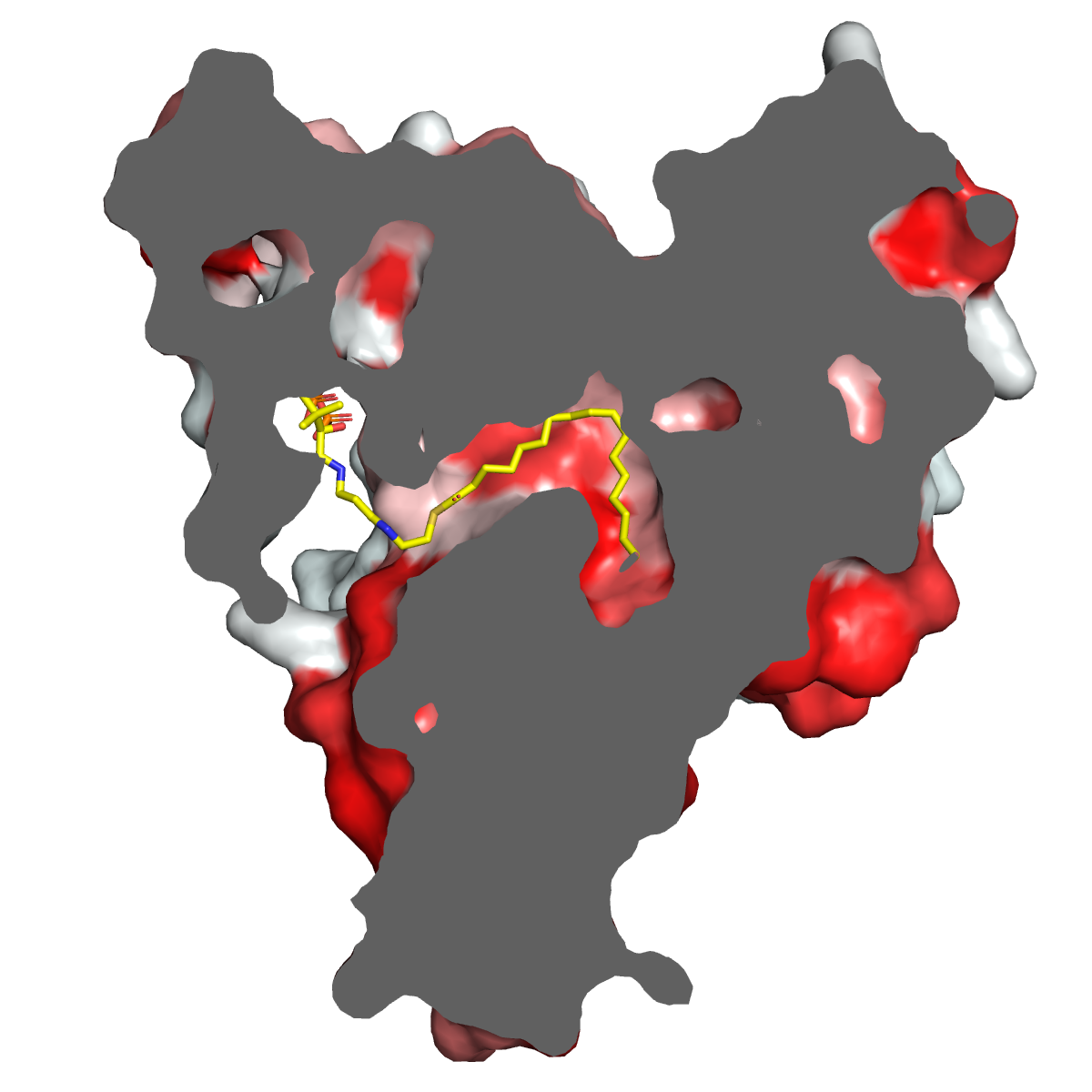
Selecciona con PyMol los aminoácidos a una distancia de 5 Armstrons de un objeto
select selcenter, (byres ligand-Xtal///STI/* around 5)
select selcenter, (byres obj-name///res-name/* around 5)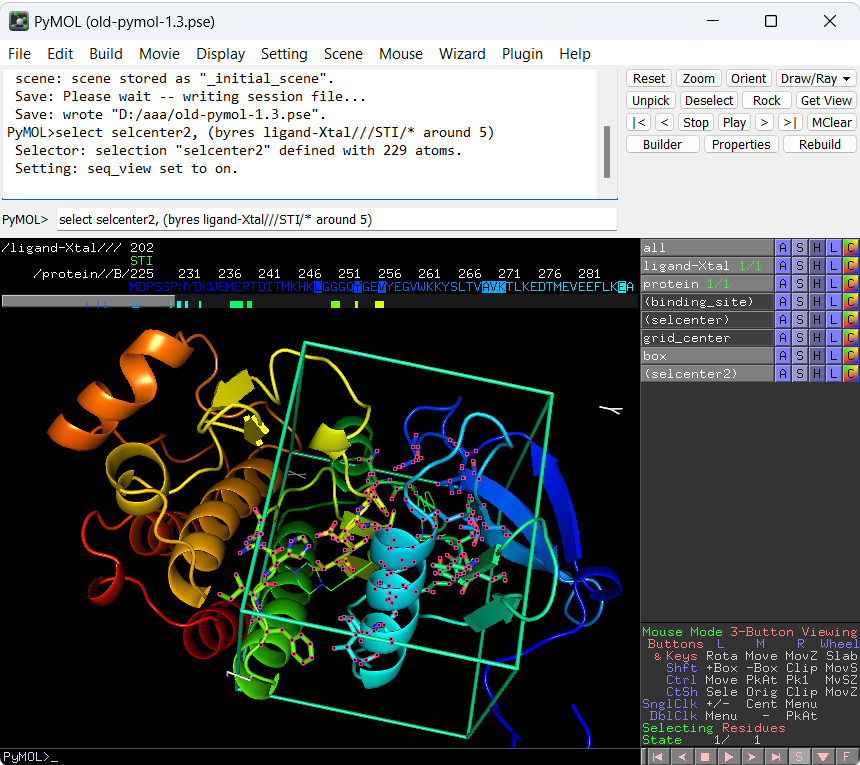
How to install Python 2.7 as an additional kernel next to the default Python 3.X one?

# install everything (except JupyterHub itself) with Python 2 and 3. Jupyter is included in Anaconda.
conda create -n py3 python=3 anaconda
conda create -n py2 python=2 anaconda
# register py2 kernel
source activate py2
ipython kernel install
# same for py3, and install juptyerhub in the py3 env
source activate py3
ipython kernel install
pip install jupyterhubCambiar nombre de una cadena/residuo de una proteína
select the chain for which you wish to add/alter the chain id
# make sure that the selecting state (on the bottom right side) is on chains
type the command: alter (chain A),chain='B'
# This will alter the chain id to B, if you want another id change the command accordingly
type the command: alter (sele),resn='INH'
# This will alter the residue id to INH, if you want another id change the command accordingly
type the command: sortSeleccionar el atom ID 4557 del obj01
select 4557, obj01 and id 4557ball and stick, "ball" or obj01
preset.ball_and_stick(selection='all', mode=1)Renumber the amino acids in a protein, so that it starts from 0 instead of its offset as defined in the structure file
alter (all),resi=str(int(resi)+100) # it starts from 100
alter (all),resi=str(int(resi)-7) # it starts from -7
# refresh (turn on seq_view to see what this command does).
sortColor of background for YASARA
ColorBG 0000FF,FFFFFF,0000FF,FFFFFF
squeue jobs para SLURN
squeue -u jant.encinar -ao "%.14i %.10u %.8j %.3C %.5m %.6b %.6Q %.5q %.10M %.7T %.15R %E" $*
squeue -u jant.encinar --sort=+j #sort by the job-name (given by #SBATCH --job-name XXX)
rm *.err *.out *.adr *clean.sce *_bestposes.pdb *_bestposes.sdf *_checkpoint.sce
LD_LIBRARY_PATH=/your/path/to/yasara:$LD_LIBRARY_PATH; export LD_LIBRARY_PATHListar, desde la linea de comandos, los archivos *.out con el comando find y después borrarlos.
sudo find / -name "default.ini" 2>/dev/null # desde consola Linux
find . -name "*.out" -type f
find . -name "*.out" -type f -delete
# Want to copy files from SSH between hosts on a network? Try the scp command:
scp file.txt jant.encinar@castleblack.umhnet.es:/path/to/dest/
scp demo.txt jant.encinar@castleblack.umhnet.es:/temp/
# You can use secure copy (scp) with the recursive option (-r):
scp -r /path/to/local/dir jant.encinar@castleblack.umhnet.es:/path/to/remote/dir
# If you want to copy a directory from machine a to b while logged into a:
scp -r /path/to/directory jant.encinar@castleblack.umhnet.es:/path/to/destination
# If you want to copy a directory from machine a to b while logged into b:
scp -r jant.encinar@castleblack.umhnet.es:/path/to/directory /path/to/destinationYASARA files: delete useless YASARA's docking files
rm *.adr *clean.sce *_checkpoint.sce *_bestposes.pdb *_bestposes.sdf *_ligands.sdfls Linux: display or list all directories in Unix
ls -d */YASARA files: delete all selected files
rm *.mcr *.tab *.yob *.png *.txt *.html *last.pdb *last.sce *average.pdb *min.pdb
*min.sce *_dccm.sce *.job *.sbatchNombre de hoja Excel: función para incluir en una celda (en el ejemplo la celda A1 y poder operar de forma lógica con ella) el nombre de cada hoja de un libro Excel.
=DERECHA(CELDA("nombrearchivo",A1),LARGO(CELDA("nombrearchivo",A1))-HALLAR("]",CELDA("nombrearchivo",A1)))Pymol: calcula una orientación determinada.
PyMOL>get_view
### cut below here and paste into script ###
set_view (\
0.387747556, -0.530695796, -0.753665924,\
-0.785253763, 0.238018751, -0.571598351,\
0.482731193, 0.813453972, -0.324439615,\
0.000000000, 0.000000000, -285.253662109,\
41.038238525, -12.994083405, 29.010128021,\
224.896163940, 345.611175537, -20.000000000 )
### cut above here and paste into script ###Pymol: repite una orientación determinada.
PyMOL>set_view (\
0.387747556, -0.530695796, -0.753665924,\
-0.785253763, 0.238018751, -0.571598351,\
0.482731193, 0.813453972, -0.324439615,\
0.000000000, 0.000000000, -285.253662109,\
41.038238525, -12.994083405, 29.010128021,\
224.896163940, 345.611175537, -20.000000000 )Pymol: selecciona los aminoácidos 15, 356, 398, 425, 430, 447, 448, 450, 451, 480, 481, 490
sele resi 15+356+398+425+430+447+448+450+451+480+481+490Pymol: selecciona todas las prolinas.
sel arginines, resn arg
select resi 91 and resn Met and chain KPymol: combina los PR01 y obj01 en un nuevo objeto llamado Pro1-B03-RS.
create Pro1-B03-RS, PR01 + obj01Pymol: selecciona los aa a menos de 5 A del aa 142.
select near142, resi 142 around 5Pymol: selecciona todos los átomos de fosforo.
select fosforo, name pPymol: tamaño de la leyenda.
set label_size, 22
set label_position,(-2,2,1)
set label_color, black, object
# combina dos objetos (hKLC-1_model y peptide) formando un tercero (object3)
create object3, hKLC-1_model + peptidePymol: localización de la leyenda.
set label_position,(3,2,1)
Offsets the labels 3 Å in X, 2 in Y and 1 in Z, relative to the Viewport.INTRANET: localización de unidades de red
\\eparadorw01.umhnet.es\shaker$
\\discodered\qy_matdocbioq$
\\discodered.umhnet.es\jant.encinarPymol: transparencia al 50% del objeto "obj01".
set transparency:0.5, obj01Pymol: Pair fit.
# superimpose protA residues 10-25 and 33-46 to protB residues 22-37 and 41-54:
pair_fit protA///10-25+33-46/CA, protB///22-37+41-54/CA
# superimpose ligA atoms C1, C2, and C4 to ligB atoms C8, C4, and C10, respectively:
pair_fit ligA////C1, ligB////C8, ligA////C2, ligB////C4, ligA////C3, ligB////C10Pymol: Run InterfaceResidues.py
1.- Open PDB file
2.- File -> Run -> InterfaceResidues.py
3.- Enter the comman line:
interfaceResidue MD2020-2059-MERS-compl-mod1, chain G, chain F
MD2020-2059-MERS-compl-mod1 -> name of object
chain G, chain F -> chain ID of two chainSyntax to rename a directory on Unix.
mv old-folder-name new-folder-name
mv /path/to/old /path/to/newContar un gran número de ficheros.
# Ejecuta el siguiente comando desde la consola y dentro del directorio en el que quieras
# contar esos ficheros. En el ejemplo contarás los ficheros *.sim
find -type f -name '*.sim' | wc -l
# Memoria libre en cada nodo, para SGE.
qstat -f -u '*' -F mem_free
# Cuando quieres listar el contenido de un directorio donde existen muchos ficheros y con ls
# aparece el argumento "Argument list too long". La solución está en el siguiente comando que
# has de modificar en función del /directorio y el tipo de archivo a buscar, en mi caso
# ".docked.pdbqt". Generará el fichero lista-hechos.txt
find /directorio -name "*.docked.pdbqt" >/directorio/lista-hechos.txt
find -type f -name '*.docked.pdbqt' >lista-hechos.txt
# Borrar un gran número de ficheros.
find . -name "*.docked.pdbqt" -print0 | xargs -0 rm
find . -name "*_bestposes.pdb" -type f -delete
find . -name "*_bestposes.sdf" -type f -delete
find . -name "*_checkpoint.sce" -type f -delete
find . -name "*.err" -type f -delete
find . -name "*.out" -type f -delete
find . -name "*.adr" -type f -deleteConvertir ficheros mol2 a pdbqt usando OpenBabel.
https://openbabel.org/docs/dev/Command-line_tools/babel.html
"C:\Program Files (x86)\ChemAxon\MarvinBeans\bin\molconvert" mol2
D:\encinar\chemical-libraries\19-2_drug_bank\_3Dstructures.sdf
-o D:\encinar\chemical-libraries\19-2_drug_bank\drug.mol2 -m -g -F