MM/GB(PB)SA Calculations for Virtual Screening.
Follow the instructions that you can read at https://github.com/dptech-corp/Uni-GBSA. Read the paper Yang et al. Uni-GBSA: an open-source and web-based automatic workflow to perform MM/GB(PB)SA calculations for virtual screening. Brief Bioinform. 2023 Jul 20;24(4):bbad218. DOI: 10.1093/bib/bbad218
Dong, L., Qu, X., Zhao, Y., & Wang, B. (2021). Prediction of binding free energy of protein-ligand complexes with a hybrid molecular mechanics/generalized born surface area and machine learning method. ACS omega, 6(48), 32938-32947.DOI: 10.1021/acsomega.1c04996
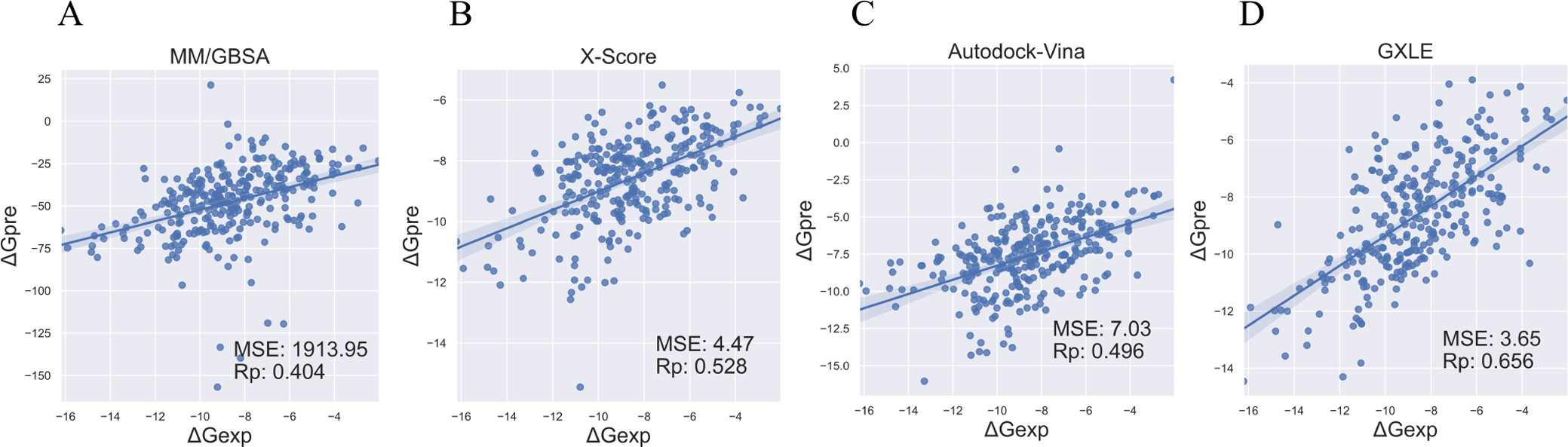
Pearson correlation coefficients and mean-squared error between the experimental data and the predicted binding free energies: (A) calculated by MM/GBSA, (B) X-Score, (C) AutoDock Vina, and (D) GXLE on the validation set.
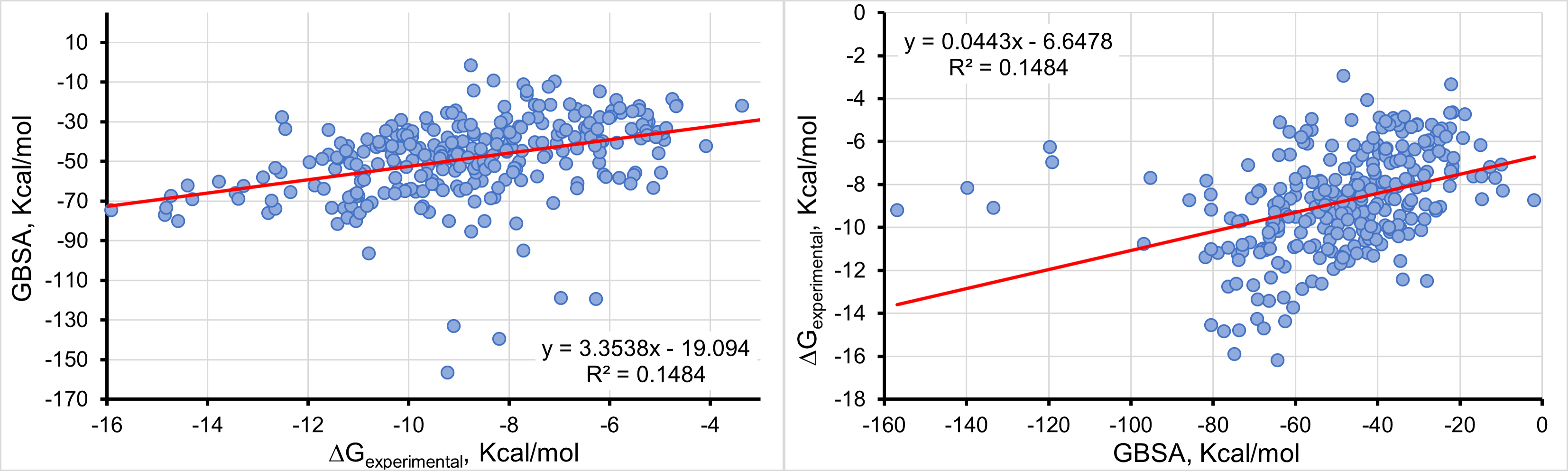
Calculate ΔGexp (Kcal/mol) as a function of GBSA, Kcal/mol
Equation 2: Y = 0.0443X - 6.4478
Equation 1: Y = 3.5338X - 19.094
Background
Calculating the binding free energy of a ligand to a protein receptor is a crucial goal in drug discovery. Molecular mechanics/Generalized-Born (Poisson-Boltzmann) surface area (MM/GB(PB)SA), which balances accuracy and efficiency, is one of the most widely used methods for evaluating ligand binding free energies in virtual screening. Uni-GBSA is an automatic workflow to perform MM/GB(PB)SA calculations. It includes several functions, including but not limited to topology preparation, structure optimization, binding free energy calculation, and parameter scanning for MM/GB(PB)SA calculations. Additionally, it has a batch mode that allows the evaluation of thousands of molecules against one protein target simultaneously, enabling its application in virtual screening.
Install on Linux as administrator
conda create -n gbsa -c conda-forge acpype openmpi mpi4py gromacs "gmx_mmpbsa>=1.5.6"
conda activate gbsa
pip install unigbsa lickitExample
unigbsa-pipeline -i 8skl-rec.pdb -l 8SKL-lig.mol2 -o BindingEnergy-8SKL.txt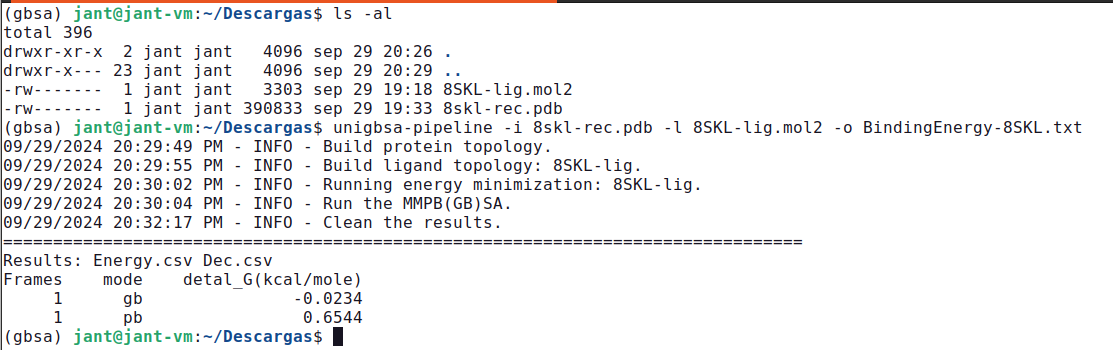
unigbsa-pipeline -i 7UAD-rec.pdb -l 7UAD-lig.mol2 -o BindingEnergy-7UAD.txt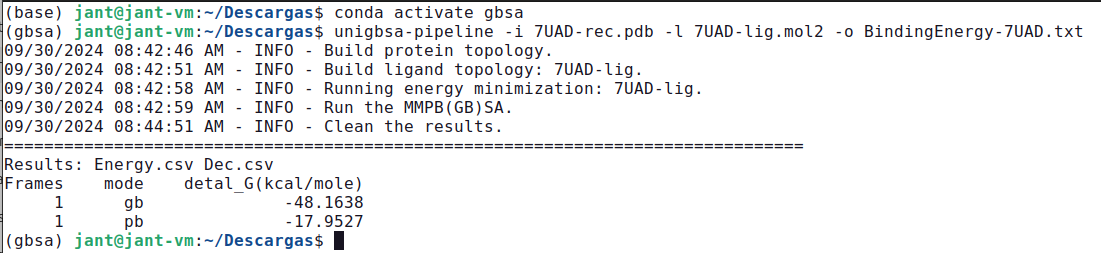
# You can update the Uni-GBSA package by command for protein-protein interaction calculation:
pip install unigbsa==0.1.7
# Run a calculation with the next example for protein-protein interaction:
unigbsa-pipeline -i MD02-1p9m-IL6-mod-control_receptor.pdb -l MD02-1p9m-IL6-mod-control_A-ligand.pdb
unigbsa-pipeline -i MD02-1p9m-IL6-mod-control_receptor.pdb -l MD02-1p9m-IL6-mod-control_C-ligand.pdb
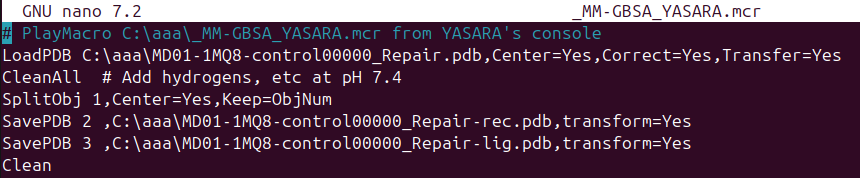
# Copy and paste the next sentence to save receptor and ligand at YASARA software:
CleanAll
SplitObj 1,Center=Yes,Keep=ObjNum
SavePDB 1 ,D:\aaa\8SKL-receptor.pdb, transform=Yes
SaveMOL2 2 ,D:\aaa\8SKL-ligand2.mol2, transform=Yes
# Run at Ubuntu 24.04 Linux:
(conda) jant@ROJO:/mnt/d/aaa$ conda activate gbsa
(gbsa) jant@ROJO:/mnt/d/aaa$ unigbsa-pipeline -i 8SKL-receptor.pdb -l 8SKL-ligand2.mol2 -o BinEn-8SKL.csv
11/02/2024 12:27:37 PM - INFO - Build protein topology.
11/02/2024 12:27:41 PM - INFO - Build ligand topology: 8SKL-ligand2.
11/02/2024 12:27:50 PM - INFO - Running energy minimization: 8SKL-ligand2.
11/02/2024 12:27:52 PM - INFO - Run the MMPB(GB)SA.
11/02/2024 12:30:25 PM - INFO - Clean the results.
================================================================================
Results: Energy.csv Dec.csv
Frames mode delta_G(kcal/mole)
1 gb -50.6667
1 pb -20.4320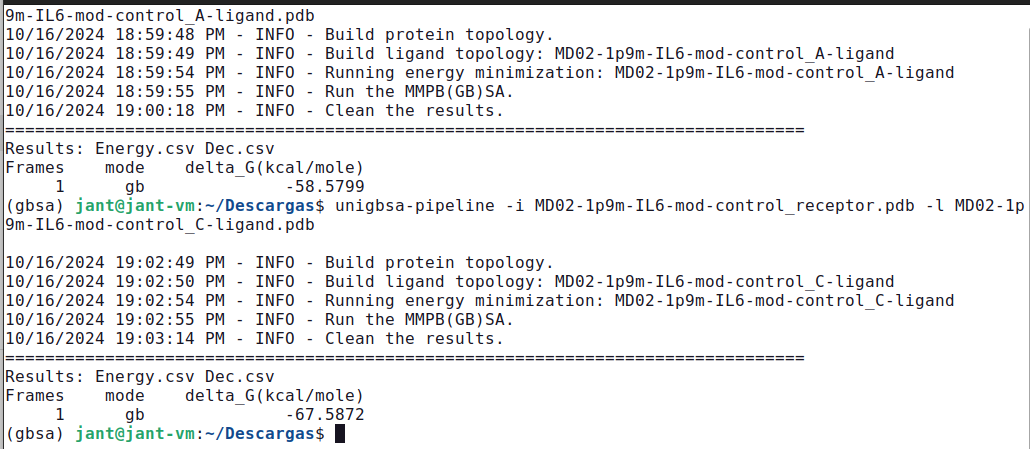
Download 8skl-rec.pdb, always with hydrogens...
Download 8skl-lig.mol2 and see last column: charge.
You can change the parameters for the MM/GB(PB)SA calculations by providing a config file (default.ini). Located at /home/jant/miniconda3/envs/gbsa/lib/python3.9/site-packages/unigbsa/data/default.ini
; parameters for simulation
[simulation]
; input pose process method:
; input - just use input pose to calculation
; em - run a simple energy minimization for the input poses
; md - run a md simulation for the input poses
mode = em
; simulation box type: triclinic, cubic, dodecahedron, octahedron
boxtype = triclinic
; Distance between the solute and the simulation box
boxsize = 0.9
; Specify salt concentration (mol/liter). This will add sufficient ions to reach up to the specified concentration
conc = 0.15
; number of md simulation steps
nsteps = 500000
; number of equilibrium simulation(nvt, npt) steps
eqsteps = 50000
; number of structure to save for the md simulation
nframe = 100
; protein forcefield (gromacs engine)
proteinforcefield = amber03
; ligand forcefield (acpype engine)
ligandforcefield = gaff
; ligand charge method: bcc, gas
ligandCharge = bcc
; parameters for PBSA/GBSA calculation, support all the gmx_MMPBSA parameters
[GBSA]
; calculation name
sys_name = GBSA
; calculation mode, Separated by commas. gb,pb,decomposition
modes = gb,pb
; best parameters for PBSA/GBSA calculations obtained from Wang, Ercheng, et al. Chemical reviews 119.16 (2019): 9478-9508.
igb = 2
indi = 4.0
exdi = 80.0The following macro to run in YASARA allows to convert a PDB with ligand and receptor into two PDBs: one for ligand and one for receptor.
import subprocess
# Ruta del archivo con la lista de nombres de PDBs (lig y rec)
ruta_lista = "/home/jant/aaa/_lista.txt"
# Ruta del archivo donde se guardará el log de salida
ruta_log = "/home/jant/aaa/salida_log.txt"
# Abrir el archivo de log en modo de escritura
with open(ruta_log, "w") as log_file:
# Leer los valores de /home/jant/aaa/_lista.txt
with open(ruta_lista, "r") as archivo:
variables = [linea.strip() for linea in archivo if linea.strip()]
# Ejecutar unigbsa-pipeline para cada variable
for variable in variables:
comando = f"unigbsa-pipeline -i {variable}_Repair-rec.pdb -l {variable}_Repair-lig.pdb -o energy-{variable}.csv"
try:
# Ejecutar el comando en la terminal y capturar la salida
resultado = subprocess.run(comando, shell=True, check=True, stdout=subprocess.PIPE, stderr=subprocess.PIPE, text=True)
# Escribir la salida del comando en el archivo log
log_file.write(f"Ejecutado con éxito para: {variable}\n")
log_file.write(resultado.stdout)
except subprocess.CalledProcessError as e:
# Escribir el error en el archivo log
log_file.write(f"Error al ejecutar el comando para: {variable}. Error:\n")
log_file.write(e.stderr)
print(f"La salida se ha guardado en {ruta_log}")# -*- coding: utf-8 -*-
import os
# Definir las rutas de entrada y salida
input_directory = '/home/jant/gbsa-51'
output_file = '/home/jant/gbsa-02/_resultados-GBSA-MD51-Orf7a-wt-IL-6_33.txt'
# Lista para almacenar los resultados
results = []
# Anadir la cabecera con comas sustituidas por tabuladores
header = "ligandName\tFrames\tmode\tcomplex\treceptor\tligand\tInternal\tVan der Waals\tElectrostatic\tPolar Solvation\tNon-Polar Solvation\tGas\tSolvation\tTOTAL\tstatus\n"
results.append(header)
# Iterar sobre los archivos en el directorio
for filename in os.listdir(input_directory):
if filename.endswith('.csv'):
file_path = os.path.join(input_directory, filename)
# Leer el archivo y extraer las lineas necesarias
with open(file_path, 'r') as file:
lines = file.readlines()
if len(lines) >= 3: # Asegurarse de que hay al menos 3 lineas
second_line = lines[1].strip().replace(',', '\t')
third_line = lines[2].strip().replace(',', '\t')
# Agregar las lineas procesadas a la lista de resultados
results.append(f"{second_line}\n{third_line}\n")
# Escribir los resultados en el archivo de salida
with open(output_file, 'w') as output:
output.writelines(results)
print(f"Datos extraidos y guardados en: {output_file}")
#!/bin/bash
#SBATCH --job-name=gbsa_02 # Numero de la simulacion de MD
#SBATCH --output=logs/output_%A_%a.out # Salida para cada tarea en el array
#SBATCH --error=logs/error_%A_%a.err # Archivo de error para cada tarea en el array
#SBATCH --array=0-999 # Rango de tareas (para 1000 valores)
#SBATCH --time=07:00:00 # Tiempo maximo por tarea
#SBATCH --cpus-per-task=2 # Numero de CPUs por tarea
#SBATCH --mem=12G # Memoria por tarea
# Cargar el entorno de conda (es necesario)
eval "$(/home/jant.encinar/miniconda3/bin/conda shell.bash hook)"
conda activate gbsa # Cambia al nombre del entorno gbsa
# Obtener el valor especifico de la lista de variables y eliminar caracteres '\r'
VARIABLE=$(sed -n "$((SLURM_ARRAY_TASK_ID + 1))p" /home/jant.encinar/test-gbsa/example_sbatch/_lista-GBSA.txt | tr -d '\r')
# Ejecutar el comando con el valor especifico
unigbsa-pipeline -i "/home/jant.encinar/test-gbsa/example_sbatch/Orf7a/${VARIABLE}_Repair-rec.pdb" \
-l "/home/jant.encinar/test-gbsa/example_sbatch/Orf7a/${VARIABLE}_Repair-lig.pdb" \
-o "/home/jant.encinar/test-gbsa/example_sbatch/Orf7a/energy-${VARIABLE}.csv"
La línea eval "$(/home/jant.encinar/miniconda3/bin/conda shell.bash hook)" se utiliza en un script de tipo sbatch para configurar el entorno conda en un entorno de clúster, como aquellos que utilizan un sistema de administración de trabajos como SLURM. Esta sintaxis permite que el script cargue y active las funcionalidades de conda en un entorno de bash (shell).
Explicación de cada parte del comando:
-
/home/jant.encinar/miniconda3/bin/conda shell.bash hook:
Este comando ejecuta el script de conda que configura las variables de entorno necesarias para que conda funcione en un entorno de bash.
shell.bash hookes una función especial de conda que genera el código necesario para configurar la shell de modo que pueda entender los comandosconda activateyconda deactivate. -
$():
El uso de
$( ... )captura la salida del comando dentro de los paréntesis y la coloca en la posición de esta expresión en el código. -
eval:
evalejecuta el resultado de la expresión. En este caso, ejecuta el código generado por el comandoconda shell.bash hook. Sineval, el código generado porconda shell.bash hooksolo se mostraría en pantalla en lugar de ser ejecutado.