MolPort database v7
Construcción de una librería de compuestos químicos comercialmente disponibles en MolPort en formato SDF y 3D destinada a simulaciones de molecular docking. A continuación se muestra scripts de Python para ejecutar en Windows, en mi caso por comodidad, desde Visial Studio Code y usando como kernel un entorno virtual creado con Miniconda3 que usa pandas, rdkit y otros módulos de Python.
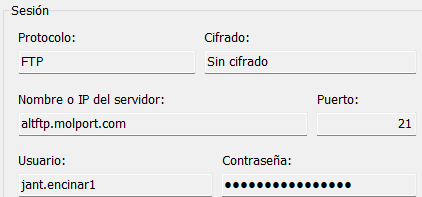
Puedes empezar por solicitar un usuario a MolPort para que te deje acceso a su servidor FTP: altftp.molport.com de forma que puedas descargar los ficheros de texto con la información de los SMILES y Molport ID de las moleculas disponibles en cada momento a la venta, que lógicamente cambian, unas aparecen nuevas y otras dejan de estar disponibles. Para la construcción de esta versión de la base de datos que describo en este post, he descargado su base de datos completa, en forma de 102 ficheros comprimidos con tar.gz, cuya descompresión libera ficheros de texto con la información que ves en la figura inferior derecha.
 |
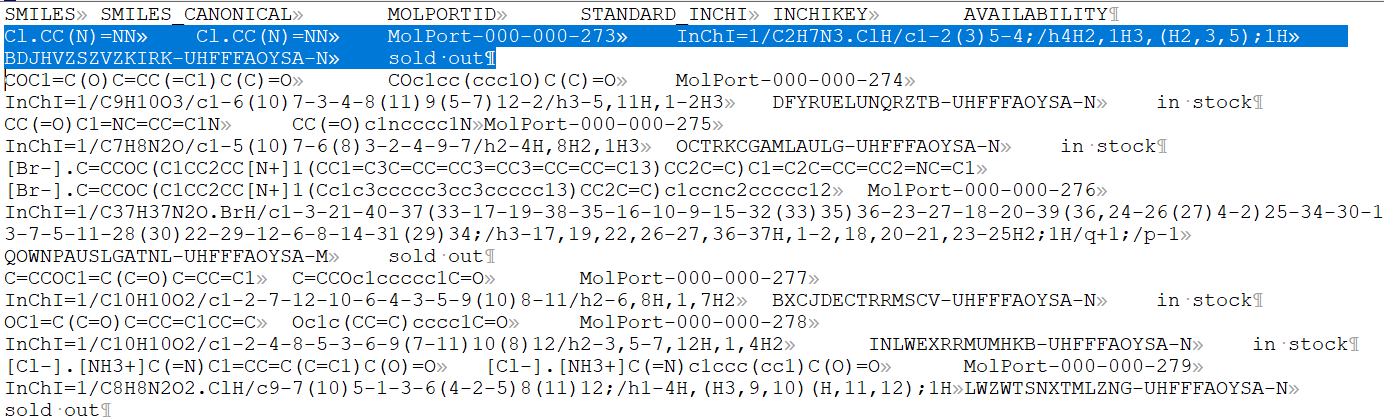 |
Cada uno de los ficheros de texto descargado de MolPort tiene información en columnas seperadas por tabuladores. La primera tarea consiste en rescatar la información de las columnas: SMILES y MOLPORTID. Será muy util en este proceso largo y complejo guardar copias de los ficheros que generamos en cada paso. Algunos procesos tardan varias horas en ejecutarse completamente en una máquina con SO Windows 11 y procesador I7. Es probable que ejecutando en Linux se agilicen los tiempos de ejecuación pero será necesario adaptar los scripts de Python que están escritos para ejecutarse en Windows.
El siguiente script de Python, para ejecutar en Windows, busca en la carpeta D:\MolPort-database_v7\ todos los ficheros de texto con extensión *.txt y rescata de cada uno de ellos los valores de las columnas "SMILES" y "MOLPORTID". Genera de salida otro fichero de texto con el mismo nombre que el fichero de entrada más la cadena "-smiles". Ejemplo de nombre de fichero de entrada: fulldb_smiles-000-000-000--000-499-999.txt y de fichero de salida: fulldb_smiles-000-000-000--000-499-999-smiles.txt
Debes instalar pandas, si aún no lo tienes, usando desde la consola CMD (Administrador) la siguiente sintaxis:
pip install pandasimport os
import pandas as pd
# Ruta de la carpeta donde se encuentran los archivos
input_folder = r"D:\MolPort-database_v7"
# Iterar sobre todos los archivos en la carpeta
for file_name in os.listdir(input_folder):
if file_name.endswith(".txt"):
input_file_path = os.path.join(input_folder, file_name)
# Leer el archivo de entrada
try:
data = pd.read_csv(input_file_path, sep="\t") # Ajustar el separador si no es tabulación
except Exception as e:
print(f"Error al leer el archivo {file_name}: {e}")
continue
# Verificar si las columnas "SMILES" y "MOLPORTID" existen
if "SMILES" in data.columns and "MOLPORTID" in data.columns:
output_data = data[["SMILES", "MOLPORTID"]]
output_file_name = f"{os.path.splitext(file_name)[0]}-smiles.txt"
output_file_path = os.path.join(input_folder, output_file_name)
# Guardar el archivo de salida
try:
output_data.to_csv(output_file_path, sep="\t", index=False)
print(f"Archivo generado: {output_file_path}")
except Exception as e:
print(f"Error al escribir el archivo {output_file_name}: {e}")
else:
print(f"El archivo {file_name} no contiene las columnas necesarias.")Este ejemplo te muestra la información que has rescatado y te permite adevertir que MolPort te deja información que será necesario eliminar pensado en que vas a usar la base de datos para simulaciones de docking y necesitas ficheros SDF en los que cada molécula sea única, luego tendremos que eliminar iones, sales, etc. Los SMILES separados por "." (punto) indican que hay más de una molécula. Mi criterio es quedarme con la cadena de texto más grande de cada línea que tenga "." (punto) y eliminar todas las demás. 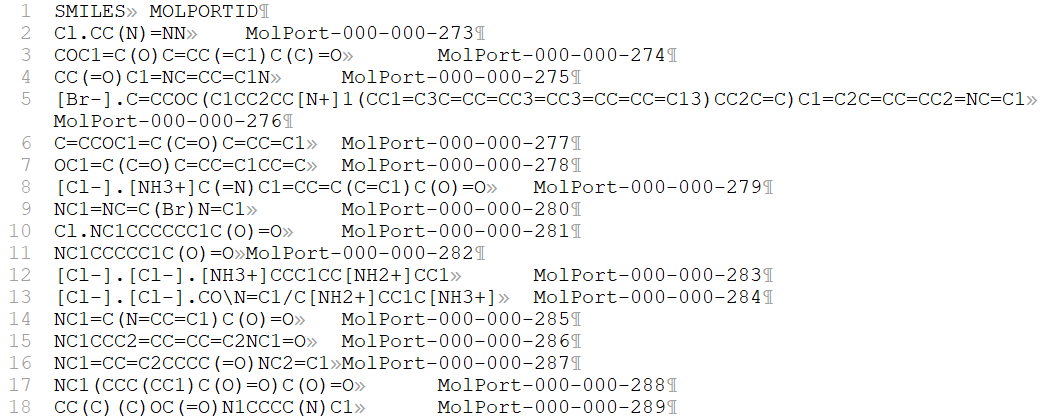
Necesito eliminar de cada fichero *-smiles.txt, generado con el script anterior, en la columna de SMILES aquellas cadenas de texto anteriores o posteriores a un punto "." La columna de SMILES tiene información que permite generar la estructura de moléculas en 3D y algunos valores de esta columna tienen más de una molécula, separadas por punto "."; por tanto, el script debe buscar en la ruta D:\MolPort-database_v7, en cada fichero cuyo nombre termine en "-smiles.txt" cadenas de texto que contengan punto "." , elimiminar todas las más pequeñas antes o después del "." y quedarse con la cadena más grande. De salida el script debe generar otro fichero de texto cuyo nombre sea el del fichero de entrada terminado en _sin-puntos.txt.
Ejemplo de nombre de fichero de entrada: fulldb_smiles-000-000-000--000-499-999-smiles.txt y de fichero de salida: fulldb_smiles-000-000-000--000-499-999_sin-puntos.txt. Vease la diferencia...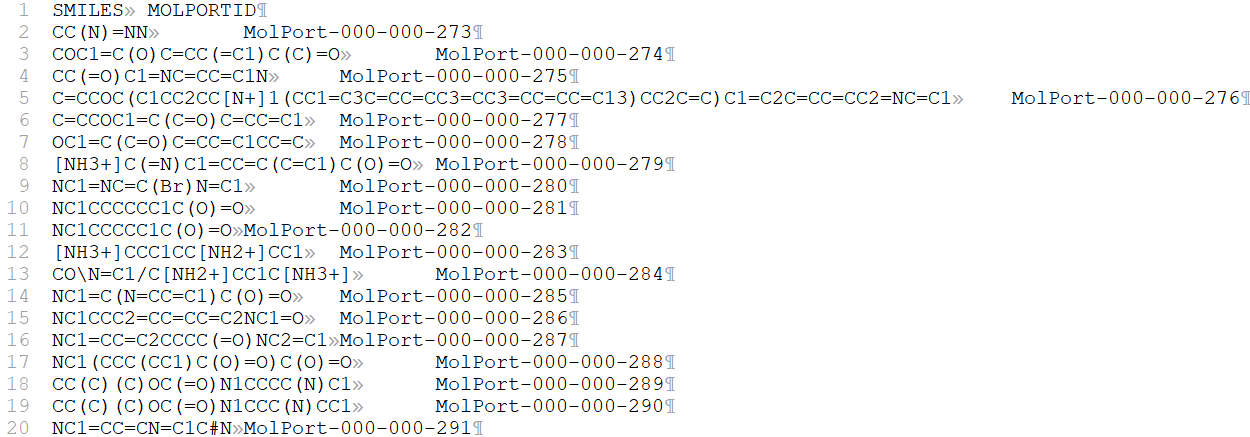
import os
import pandas as pd
# Ruta de la carpeta donde se encuentran los archivos
input_folder = r"D:\MolPort-database_v7"
# Iterar sobre todos los archivos en la carpeta
for file_name in os.listdir(input_folder):
if file_name.endswith("-smiles.txt"):
input_file_path = os.path.join(input_folder, file_name)
try:
# Leer el archivo de entrada
data = pd.read_csv(input_file_path, sep="\t")
except Exception as e:
print(f"Error al leer el archivo {file_name}: {e}")
continue
# Verificar si la columna "SMILES" existe
if "SMILES" in data.columns:
smiles_column = data["SMILES"]
processed_smiles = []
removed_smiles = []
for entry in smiles_column:
if "." in entry:
# Dividir las cadenas por el punto y seleccionar la más larga
parts = entry.split(".")
largest_part = max(parts, key=len)
removed_parts = [part for part in parts if part != largest_part]
processed_smiles.append(largest_part)
removed_smiles.append(".".join(removed_parts))
else:
processed_smiles.append(entry)
removed_smiles.append("")
# Crear los DataFrames de salida
data["SMILES"] = processed_smiles
removed_data = pd.DataFrame({"SMILES Eliminado": removed_smiles})
# Generar nombres de archivo de salida
output_processed_file = os.path.join(input_folder, file_name.replace("-smiles.txt", "_sin-puntos.txt"))
output_removed_file = os.path.join(input_folder, file_name.replace("-smiles.txt", "_eliminado.txt"))
# Guardar los archivos de salida
try:
data.to_csv(output_processed_file, sep="\t", index=False)
removed_data.to_csv(output_removed_file, sep="\t", index=False)
print(f"Archivos generados: {output_processed_file}, {output_removed_file}")
except Exception as e:
print(f"Error al escribir archivos de salida para {file_name}: {e}")
else:
print(f"El archivo {file_name} no contiene la columna 'SMILES'.")Necesitamos filtar esta libreria "monstruosa" y reducir el número de moléculas disponibles para simulaciones de molecular docking y para ello vamos a usar varios desciptores de esas moléculas que nos permitan descartar aquellas moléculas que no cumplan los rangos de valores de esos descriptores. Podemos tener en cuenta las reglas de Lipinski.
El siguiente script de Python, para ejecutar en Windows, busca en la ruta D:\MolPort-database_v7 todos los ficheros de texto cuyo nombre termine en _sin-puntos.txt y en los que en la primera columna encuentre SMILES y en la segunda MOLPORTID, para calcular el peso molecular de la molécula a la que da lugar cada smiles, los valores de logP, topological polar surface area: TPSA y número de enlaces rotables: RotatableBonds. Una vez calculados estos parámetros los colocará en columnas sucesivas, generando un nuevo fichero de salida cuyo nombre sea el del fichero de entrada añadiendo _MW_logP_TPSA_RotatableBonds.txt.
import os
import pandas as pd
from rdkit import Chem
from rdkit.Chem import Descriptors
# Ruta de la carpeta donde se encuentran los archivos
input_folder = r"D:\MolPort-database_v7"
# Iterar sobre todos los archivos en la carpeta
for file_name in os.listdir(input_folder):
if file_name.endswith("_sin-puntos.txt"):
input_file_path = os.path.join(input_folder, file_name)
try:
# Leer el archivo de entrada
data = pd.read_csv(input_file_path, sep="\t")
except Exception as e:
print(f"Error al leer el archivo {file_name}: {e}")
continue
# Verificar si las columnas "SMILES" y "MOLPORTID" existen
if "SMILES" in data.columns and "MOLPORTID" in data.columns:
molecular_weights = []
logP_values = []
tpsa_values = []
# num_atoms_values = []
rotatable_bonds_values = []
for smiles in data["SMILES"]:
try:
# Convertir SMILES a una molécula RDKit
mol = Chem.MolFromSmiles(smiles)
if mol:
# Calcular descriptores
mw = Descriptors.MolWt(mol)
logP = Descriptors.MolLogP(mol)
tpsa = Descriptors.TPSA(mol)
rotatable_bonds = Descriptors.NumRotatableBonds(mol)
else:
mw = logP = tpsa = rotatable_bonds = None
except Exception as e:
print(f"Error al procesar SMILES {smiles} en archivo {file_name}: {e}")
mw = logP = tpsa = rotatable_bonds = None
molecular_weights.append(mw)
logP_values.append(logP)
tpsa_values.append(tpsa)
rotatable_bonds_values.append(rotatable_bonds)
# Agregar las columnas calculadas al DataFrame
data["MW"] = molecular_weights
data["logP"] = logP_values
data["TPSA"] = tpsa_values
data["RotatableBonds"] = rotatable_bonds_values
# Generar nombre de archivo de salida
output_file_path = os.path.join(input_folder, file_name.replace("_sin-puntos.txt", "_MW_logP_TPSA_RotatableBonds.txt"))
# Guardar el archivo de salida
try:
data.to_csv(output_file_path, sep="\t", index=False)
print(f"Archivo generado: {output_file_path}")
except Exception as e:
print(f"Error al escribir el archivo de salida para {file_name}: {e}")
else:
print(f"El archivo {file_name} no contiene las columnas necesarias 'SMILES' y 'MOLPORTID'.")
Cuando ya disponga de los ficheros de texto con los cuatro parámetros calculados: MW, logP, TPSA y RotatableBonds usaré el siguiente script de Python para filtrar compuestos con MW entre 300-750, logP menor o igual a 3, TPSA menor o igual a 140 y RotatableBonds menor o igual a 7.
El siguiente script de Python para ejecutar en Windows, lee todos los ficheros de texto en la ruta D:\MolPort-database_v7, cuyo nombre termine en "_MW_logP_TPSA_RotatableBonds.txt", que contienen seis columnas y en la primera columna tienen el valor de SMILES, en la tercera columna el peso molecular (MW), en la cuarta columna tiene el valor de logP, en la quinta columna tiene el valor de TPSA y detecta las moléculas con peso molecular mayor o igual a 300 y menor de 750, que además tengan un valor de logP menor o igual a 3, un valor de TPSA menor de 140 y 7 o menos enlaces rotables; generando un fichero de salida cuyo nombre sea el nombre el fichero de entrada sustituyendo "_MW_logP_TPSA_RotatableBonds.txt" por "_300-750_menor3_menor140_RotBonds7.txt"
import os
import pandas as pd
# Ruta de la carpeta donde se encuentran los archivos
input_folder = r"D:\MolPort-database_v7"
# Iterar sobre todos los archivos en la carpeta
for file_name in os.listdir(input_folder):
if file_name.endswith("_MW_logP_TPSA_RotatableBonds.txt"):
input_file_path = os.path.join(input_folder, file_name)
try:
# Leer el archivo de entrada
data = pd.read_csv(input_file_path, sep="\t")
except Exception as e:
print(f"Error al leer el archivo {file_name}: {e}")
continue
# Verificar si las columnas necesarias existen
required_columns = ["SMILES", "MW", "logP", "TPSA", "RotatableBonds"]
if all(column in data.columns for column in required_columns):
try:
# Aplicar filtros
filtered_data = data[
(data["MW"] >= 300) & (data["MW"] < 750) &
(data["logP"] <= 3) &
(data["TPSA"] < 140) &
(data["RotatableBonds"] <= 7)
]
# Verificar si hay datos filtrados
if not filtered_data.empty:
# Generar nombre de archivo de salida
output_file_path = os.path.join(
input_folder,
file_name.replace("_MW_logP_TPSA_RotatableBonds.txt", "_300-750_menor3_menor140_RotBonds7.txt")
)
# Guardar el archivo de salida
filtered_data.to_csv(output_file_path, sep="\t", index=False)
print(f"Archivo generado: {output_file_path}")
else:
print(f"No se encontraron moléculas que cumplan los criterios en {file_name}.")
except Exception as e:
print(f"Error al filtrar o guardar datos para {file_name}: {e}")
else:
print(f"El archivo {file_name} no contiene las columnas necesarias {required_columns}.")Las moléculas con carbonos asimétricos (4 sustituyentes diferentes o sea, con centros quirales) son un "dolor" en química médica... voy a separar las moléculas que llevo filtradas hasta este punto del proceso en "quirales" y "no quirales", de las primeras mi base de datos sólo contiene un estereosiómero de todos los posibles.
El siguiente script de Python para ejecutar en Windows, lee todos los ficheros de texto en la ruta D:\MolPort-database_v7 cuyo nombre termine en "_300-750_menor3_menor140_RotBonds7.txt" que contienen seis columnas y en la primera columna tienen el valor de SMILES, en la tercera columna el peso molecular (MW), en la cuarta columna tiene el valor de logP, en la quinta columna tiene el valor de TPSA y detecta las moléculas no quirales; generando un fichero de salida cuyo nombre sea el nombre el fichero de entrada en el que se sustituye "_300-750_menor3_menor140_RotBonds7.txt" por "_quiral.txt".
import os
import pandas as pd
from rdkit import Chem
# Ruta de entrada
directory = r"D:\\MolPort-database_v7"
# Función para detectar carbonos quirales
def detect_chiral(smiles):
try:
mol = Chem.MolFromSmiles(smiles)
if mol:
chiral_centers = Chem.FindMolChiralCenters(mol, includeUnassigned=True)
return len(chiral_centers) > 0
return False
except Exception as e:
print(f"Error procesando SMILES: {smiles}, {e}")
return False
# Procesar los archivos en la carpeta
for filename in os.listdir(directory):
if filename.endswith("_300-750_menor3_menor140_RotBonds7.txt"):
filepath = os.path.join(directory, filename)
# Leer el archivo en un DataFrame
try:
data = pd.read_csv(filepath, sep="\t", header=0)
# Comprobar que tiene las columnas esperadas
if "SMILES" in data.columns and "MOLPORTID" in data.columns:
# Filtrar moléculas quirales
chiral_molecules = data[data["SMILES"].apply(detect_chiral)]
# Generar el nombre del archivo de salida
output_filepath = os.path.join(directory, filename.replace("_300-750_menor3_menor140_RotBonds7.txt", "_quiral.txt"))
# Guardar el resultado en un nuevo archivo
if not chiral_molecules.empty:
chiral_molecules.to_csv(output_filepath, sep="\t", index=False)
print(f"Archivo generado: {output_filepath}")
else:
print(f"No se encontraron moléculas quirales en: {filename}")
else:
print(f"Archivo {filename} no contiene las columnas esperadas.")
except Exception as e:
print(f"Error procesando el archivo {filename}: {e}")El script equivalente al anterior para detectar las moléculas no quirales.
import os
import pandas as pd
from rdkit import Chem
# Ruta de la carpeta donde se encuentran los archivos
input_folder = r"D:\MolPort-database_v7"
# Iterar sobre todos los archivos en la carpeta
for file_name in os.listdir(input_folder):
if file_name.endswith("_300-750_menor3_menor140_RotBonds7.txt"):
input_file_path = os.path.join(input_folder, file_name)
try:
# Leer el archivo de entrada
data = pd.read_csv(input_file_path, sep="\t")
except Exception as e:
print(f"Error al leer el archivo {file_name}: {e}")
continue
# Verificar si la columna "SMILES" existe
if "SMILES" in data.columns:
non_chiral_rows = []
for _, row in data.iterrows():
smiles = row["SMILES"]
try:
# Convertir SMILES a una molécula RDKit
mol = Chem.MolFromSmiles(smiles)
if mol:
# Detectar si no tiene carbonos quirales
if not Chem.FindMolChiralCenters(mol, includeUnassigned=True):
non_chiral_rows.append(row)
except Exception as e:
print(f"Error al procesar SMILES {smiles} en archivo {file_name}: {e}")
# Crear un DataFrame con las filas no quirales
non_chiral_data = pd.DataFrame(non_chiral_rows)
# Generar el nombre del archivo de salida
output_file_path = os.path.join(input_folder, file_name.replace("_300-750_menor3_menor140_RotBonds7.txt", "_NO-quirales.txt"))
# Guardar el archivo de salida
try:
non_chiral_data.to_csv(output_file_path, sep="\t", index=False)
print(f"Archivo generado: {output_file_path}")
except Exception as e:
print(f"Error al escribir el archivo de salida para {file_name}: {e}")
else:
print(f"El archivo {file_name} no contiene la columna necesaria 'SMILES'.")Para hacer simulaciones de acoplamiento molecular (docking) usando el software de YASARA necesitamos ficheros SDF que no contengan más de 3000 moléculas (pueden usarse ficheros con mayor número de moléculas pero la probabilidad de que el proceso se rompa es alta).
El siguiente script de Python lee en la ruta D:\MolPort-database_v7\07__filter_300-750_menor3_menor140_RotBonds7__kirales todos los ficheros cuyo nombre termine en _quiral.txt los divide en tantos ficheros de 3000 moléculas como sea necesario y los guarda, en la ruta D:\MolPort-database_v7\09_MP7_quiral_3000\, con las columnas SMILES y MOLPORTID.
import os
import pandas as pd
# Ruta de entrada y salida
input_folder = r"D:\MolPort-database_v7\07__filter_300-750_menor3_menor140_RotBonds7__kirales"
output_folder = r"D:\MolPort-database_v7\09_MP7_quiral_3000"
# Crear la carpeta de salida si no existe
os.makedirs(output_folder, exist_ok=True)
# Límite de moléculas por archivo
LIMIT = 3000
# Contador para los archivos de salida
file_counter = 1
# Buffer para acumular datos
accumulated_data = []
# Formatear el nombre del archivo de salida
def generate_output_filename(counter):
return os.path.join(output_folder, f"MP7-Q-{counter:04d}.txt")
# Función para guardar el buffer en un archivo
def save_to_file(data, counter):
output_file = generate_output_filename(counter)
with open(output_file, "w") as f:
for row in data:
f.write("\t".join(row) + "\n")
print(f"Archivo generado: {output_file}")
# Procesar los archivos de entrada
for file_name in os.listdir(input_folder):
if file_name.endswith("_quiral.txt"):
input_file_path = os.path.join(input_folder, file_name)
try:
# Leer el archivo de entrada
data = pd.read_csv(input_file_path, sep="\t")
except Exception as e:
print(f"Error al leer el archivo {file_name}: {e}")
continue
# Verificar si las columnas SMILES y MOLPORTID existen
if "SMILES" in data.columns and "MOLPORTID" in data.columns:
for _, row in data.iterrows():
accumulated_data.append((row["SMILES"], row["MOLPORTID"]))
# Si alcanzamos el límite, guardar y resetear el buffer
if len(accumulated_data) >= LIMIT:
save_to_file(accumulated_data, file_counter)
file_counter += 1
accumulated_data = []
else:
print(f"El archivo {file_name} no contiene las columnas necesarias 'SMILES' y 'MOLPORTID'.")
# Guardar los datos restantes si hay alguno
if accumulated_data:
save_to_file(accumulated_data, file_counter)El siguiente script de Python lee en la ruta D:\MolPort-database_v7\08__filter_300-750_menor3_menor140_RotBonds7__NO-kirales todos los ficheros cuyo nombre termine en _NO-quirales.txt los divide en tantos ficheros de 3000 moléculas como sea necesario y los guarda, en la ruta D:\MolPort-database_v7\10_MP7_no-quiral_3000\, con las columnas SMILES y MOLPORTID.
import os
import pandas as pd
# Ruta de entrada y salida
input_folder = r"D:\MolPort-database_v7\08__filter_300-750_menor3_menor140_RotBonds7__NO-kirales"
output_folder = r"D:\MolPort-database_v7\10_MP7_no-quiral_3000"
# Crear la carpeta de salida si no existe
os.makedirs(output_folder, exist_ok=True)
# Límite de moléculas por archivo
LIMIT = 3000
# Contador para los archivos de salida
file_counter = 1
# Buffer para acumular datos
accumulated_data = []
# Formatear el nombre del archivo de salida
def generate_output_filename(counter):
return os.path.join(output_folder, f"MP7-noQ-{counter:04d}.txt")
# Función para guardar el buffer en un archivo
def save_to_file(data, counter):
output_file = generate_output_filename(counter)
with open(output_file, "w") as f:
for row in data:
f.write("\t".join(row) + "\n")
print(f"Archivo generado: {output_file}")
# Procesar los archivos de entrada
for file_name in os.listdir(input_folder):
if file_name.endswith("_NO-quirales.txt"):
input_file_path = os.path.join(input_folder, file_name)
try:
# Leer el archivo de entrada
data = pd.read_csv(input_file_path, sep="\t")
except Exception as e:
print(f"Error al leer el archivo {file_name}: {e}")
continue
# Verificar si las columnas SMILES y MOLPORTID existen
if "SMILES" in data.columns and "MOLPORTID" in data.columns:
for _, row in data.iterrows():
accumulated_data.append((row["SMILES"], row["MOLPORTID"]))
# Si alcanzamos el límite, guardar y resetear el buffer
if len(accumulated_data) >= LIMIT:
save_to_file(accumulated_data, file_counter)
file_counter += 1
accumulated_data = []
else:
print(f"El archivo {file_name} no contiene las columnas necesarias 'SMILES' y 'MOLPORTID'.")
# Guardar los datos restantes si hay alguno
if accumulated_data:
save_to_file(accumulated_data, file_counter)Ahora viene cuando observamos los números de vértigo: en la sección quirales tenemos 1678 ficheros de 3000 moléculas cada uno y 1 fichero con 2418 moléculas, todos ellos suman 5.0336.418 moléculas. En la sección no quirales tenemos 1633 ficheros de 3000 moléculas cada uno y 1 fichero con 1062 moléculas, todos ellos suman 4.900.062 moléculas.
En la sección no quirales tenemos 1633 ficheros de 3000 moléculas cada uno y 1 fichero con 1062 moléculas, todos ellos suman 4.900.062 moléculas.

Nuestra última tarea consiste en convertir esas 5.0336.418 + 4.900.062 moléculas en estructuras 3D protonadas, por ejemplo a pH 7.4 usando un script de YASARA.
libname='MP7-Q-0001'
Clear
for line in file (libname).txt
# Extraer SMILES y nombre
smiles,name=split line
# Construir SMILES
obj = BuildSMILES '(smiles)'
# Minimizar con NOVA para cerrar los enlaces largos que BuildSMILES dejó
# (La optimización QM no sabe dónde deben estar los enlaces)
Cell Auto,Extension=10
ph=7.4
ForceField NOVA,SetPar=yes
Experiment Minimization
Experiment On
Wait ExpEnd
# Optimizar con MOPAC para afinar la geometría
OptimizeObj (obj)
# Establecer el nombre del compuesto
CompoundMol Obj (obj),'(name)'
RemoveObj (obj)
# Guardar un único archivo SDF
AddObj all
SaveSDF !SimCell,(libname)Pero YASARA es un poco lento ... y me parece mejor idea usar RDkit para conseguir convertir los ficheros de texto con SMILES y MOLPORTID en ficheros SDF con moléculas en 3D y utilizar MMFF94 como método de optimización geométrica. Para eso sirve el siguiente script de Python de ejecución en Windows y con los módulos pandas, numpy, RDkit y tqdm. El script descarta en la salida moléculas que contienen átomos no soportados antes de optimizar la geometría. Sólo las moléculas válidas se procesarán normalmente con hidrógenos añadidos y geometrías optimizadas.
import os
from rdkit import Chem
from rdkit.Chem import AllChem
from tqdm import tqdm
# Ruta de entrada y salida
ruta_entrada = r"D:\aaa"
ruta_salida = r"D:\aaa\salida_sdf"
# Crear la carpeta de salida si no existe
os.makedirs(ruta_salida, exist_ok=True)
# Función para optimizar geometría con MMFF94
def optimizar_con_mmff94(molecula):
try:
# Generar propiedades del campo de fuerza MMFF
props = AllChem.MMFFGetMoleculeProperties(molecula)
if props is None:
return None # Si no se pueden generar propiedades, devolver None
# Crear el campo de fuerza y optimizar
ff = AllChem.MMFFGetMoleculeForceField(molecula, props)
if ff is not None:
ff.Minimize()
return molecula
return None
except Exception as e:
print(f"Error durante la optimización con MMFF94: {e}")
return None
# Iterar sobre los archivos de la ruta de entrada
for archivo in os.listdir(ruta_entrada):
if archivo.endswith(".txt"):
ruta_archivo = os.path.join(ruta_entrada, archivo)
with open(ruta_archivo, 'r') as f:
lineas = f.readlines()
# Crear una lista para almacenar moléculas optimizadas
moleculas_optimizadas = []
print(f"Procesando archivo: {archivo}")
# Crear barra de progreso
for linea in tqdm(lineas, desc=f"Procesando moléculas en {archivo}", unit="mol"):
try:
smiles, nombre = linea.strip().split('\t')
molecula = Chem.MolFromSmiles(smiles)
if molecula is not None:
# Añadir hidrógenos
molecula = Chem.AddHs(molecula)
# Generar coordenadas 3D iniciales
if AllChem.EmbedMolecule(molecula, randomSeed=42) == 0:
# Optimizar con MMFF94
molecula_optimizada = optimizar_con_mmff94(molecula)
if molecula_optimizada is not None:
molecula_optimizada.SetProp("_Name", nombre)
moleculas_optimizadas.append(molecula_optimizada)
else:
print(f"No se pudo generar la molécula a partir de SMILES: {smiles}")
except Exception as e:
print(f"Error procesando la molécula {nombre} ({smiles}): {e}")
continue
# Escribir las moléculas optimizadas en formato SDF
if moleculas_optimizadas:
nombre_salida = os.path.splitext(archivo)[0] + ".sdf"
ruta_salida_archivo = os.path.join(ruta_salida, nombre_salida)
with Chem.SDWriter(ruta_salida_archivo) as escritor:
for mol in moleculas_optimizadas:
escritor.write(mol)
print(f"Procesado: {archivo}, moléculas optimizadas guardadas en {ruta_salida_archivo}")
El siguiente script de Python, para ejecutar en Linux, usa una lista contenida en la ruta /home/jant.encinar/py-linux/36_smiles-3D-sdf/lista_01.txt, con el nombre de los ficheros de texto en columnas separadas por tabulador, que contienen en la primera columna SMILES y en la segunda MOLPORTID, localizados en la misma ruta, para realizar la operación de conversión de SMILES a ficheros SDF con moléculas en 3D y utilizar MMFF94 como método de optimización geométrica.
# -*- coding: iso-8859-1 -*-
import os
from rdkit import Chem
from rdkit.Chem import AllChem
from tqdm import tqdm
# Rutas de entrada y salida
ruta_lista_archivos = "/home/jant.encinar/py-linux/36_smiles-3D-sdf/lista_01.txt"
ruta_salida = "/home/jant.encinar/py-linux/36_smiles-3D-sdf/salida_sdf"
# Crear la carpeta de salida si no existe
os.makedirs(ruta_salida, exist_ok=True)
# Función para optimizar geometría con MMFF94
def optimizar_con_mmff94(molecula):
try:
# Generar propiedades del campo de fuerza MMFF
props = AllChem.MMFFGetMoleculeProperties(molecula)
if props is None:
return None # Si no se pueden generar propiedades, devolver None
# Crear el campo de fuerza y optimizar
ff = AllChem.MMFFGetMoleculeForceField(molecula, props)
if ff is not None:
ff.Minimize()
return molecula
return None
except Exception as e:
print(f"Error durante la optimización con MMFF94: {e}")
return None
# Leer la lista de archivos
try:
with open(ruta_lista_archivos, 'r') as f:
lista_archivos = [linea.strip() for linea in f.readlines() if linea.strip()]
except FileNotFoundError:
print(f"No se encontró el archivo de lista: {ruta_lista_archivos}")
exit(1)
# Procesar cada archivo en la lista
for archivo in lista_archivos:
ruta_archivo = f"/home/jant.encinar/py-linux/36_smiles-3D-sdf/{archivo}.txt"
if not os.path.exists(ruta_archivo):
print(f"Archivo no encontrado: {ruta_archivo}")
continue
with open(ruta_archivo, 'r') as f:
lineas = f.readlines()
# Crear una lista para almacenar moléculas optimizadas
moleculas_optimizadas = []
print(f"Procesando archivo: {archivo}.txt")
# Crear barra de progreso
for linea in tqdm(lineas, desc=f"Procesando moléculas en {archivo}.txt", unit="mol"):
try:
smiles, nombre = linea.strip().split('\t')
molecula = Chem.MolFromSmiles(smiles)
if molecula is not None:
# Añadir hidrógenos
molecula = Chem.AddHs(molecula)
# Generar coordenadas 3D iniciales
if AllChem.EmbedMolecule(molecula, randomSeed=42) == 0:
# Optimizar con MMFF94
molecula_optimizada = optimizar_con_mmff94(molecula)
if molecula_optimizada is not None:
molecula_optimizada.SetProp("_Name", nombre)
moleculas_optimizadas.append(molecula_optimizada)
else:
print(f"No se pudo generar la molécula a partir de SMILES: {smiles}")
except Exception as e:
print(f"Error procesando la molécula {nombre} ({smiles}): {e}")
continue
# Escribir las moléculas optimizadas en formato SDF
if moleculas_optimizadas:
nombre_salida = f"{archivo}.sdf"
ruta_salida_archivo = os.path.join(ruta_salida, nombre_salida)
with Chem.SDWriter(ruta_salida_archivo) as escritor:
for mol in moleculas_optimizadas:
escritor.write(mol)
print(f"Procesado: {archivo}.txt, moléculas optimizadas guardadas en {ruta_salida_archivo}")
Antes de usar el script anterior, activa el entorno virtual (en mi caso se llama jant-molport) y ejecuta el script verifica.py para asegurarte de que tienen instalado pandas, numpy, tqdm y rdkit.
(base) [jant.encinar@castleblack 36_smiles-3D-sdf]$ conda install -c conda-forge rdkit
(base) [jant.encinar@castleblack 36_smiles-3D-sdf]$ conda update -n base -c conda-forge conda
(base) [jant.encinar@castleblack 36_smiles-3D-sdf]$ conda install -c conda-forge tqdm
(base) [jant.encinar@castleblack 36_smiles-3D-sdf]$ conda install pandas numpy
(base) [jant.encinar@castleblack 36_smiles-3D-sdf]$ conda activate jant-molport
(jant-molport) [jant.encinar@castleblack 36_smiles-3D-sdf]$ python3 verifica.py
Version instalada de pandas:
2.2.3
Version instalada de numpy:
2.2.0
Version instalada de tqdm:
4.67.1
Version instalada de RDkit:
2024.09.3
(jant-molport) [jant.encinar@castleblack 36_smiles-3D-sdf]$Script verifica.py justo debajo...
import pandas as pd
import numpy as np
import tqdm
print("Version instalada de pandas")
print(pd.__version__)
print("Version instalada de numpy")
print(np.__version__)
print("Version instalada de tqdm")
print(tqdm.__version__)
from rdkit import rdBase
print("Version instalada de RDkit")
print(rdBase.rdkitVersion)Con el siguiente script de Python para ejecutar en Windows vamos a contar el número de moléculas de nuestra libreria, tanto para cada fichero *.sdf como el total.
import os
from tqdm import tqdm # Para la barra de progreso
# Ruta donde se encuentran los archivos *.sdf
ruta_sdf = r"D:\MolPort-database_v7\11_MP7_quiral_sdf"
# Archivo de salida
archivo_salida = r"D:\MolPort-database_v7\11_MP7_quiral_sdf\_lista-sdf-quiral.txt"
def contar_moleculas_en_sdf(ruta_archivo):
"""Cuenta el número de moléculas en un archivo .sdf usando $$$$ como delimitador."""
try:
with open(ruta_archivo, 'r') as archivo:
contenido = archivo.read()
# Contar ocurrencias del delimitador $$$$
return contenido.count("$$$$")
except Exception as e:
print(f"Error leyendo {ruta_archivo}: {e}")
return 0
def procesar_archivos_sdf(ruta, salida):
"""Procesa todos los archivos .sdf en la ruta dada y genera un reporte."""
try:
# Lista de archivos en la ruta
archivos_sdf = [f for f in os.listdir(ruta) if f.endswith('.sdf')]
total_archivos = len(archivos_sdf)
# Verificar si hay archivos para procesar
if total_archivos == 0:
print("No se encontraron archivos .sdf en la ruta especificada.")
return
# Inicializar contador de moléculas totales
total_moleculas = 0
# Abrir archivo de salida para escribir resultados
with open(salida, 'w') as salida_file:
# Escribir encabezado
salida_file.write("Fichero\tNumero_de_moleculas\n")
# Procesar cada archivo .sdf con barra de progreso
for archivo in tqdm(archivos_sdf, desc="Procesando archivos", unit="archivo"):
ruta_completa = os.path.join(ruta, archivo)
num_moleculas = contar_moleculas_en_sdf(ruta_completa)
total_moleculas += num_moleculas
salida_file.write(f"{archivo}\t{num_moleculas}\n")
# Escribir total de moléculas al final del archivo
salida_file.write(f"\nTOTAL\t{total_moleculas}\n")
# Mostrar total en consola
print(f"\nProcesamiento completado. Total de moléculas: {total_moleculas}")
print(f"Reporte generado en: {salida}")
except Exception as e:
print(f"Error procesando archivos en {ruta}: {e}")
# Ejecutar la función principal
if __name__ == "__main__":
procesar_archivos_sdf(ruta_sdf, archivo_salida)


El siguiente script de Python crea tantos isómeros como sea posible de las moléculas contenidas en ficheros MP7-Q-****.sdf y, en la misma ruta, crea un fichero de salida con el mismo nombre que el del fichero de entrada pero añadiendo _isomeros.sdf.
import os
from rdkit import Chem
from rdkit.Chem import AllChem
from rdkit.Chem.EnumerateStereoisomers import EnumerateStereoisomers, StereoEnumerationOptions
from tqdm import tqdm
# Ruta donde se encuentran los archivos .sdf
ruta_sdf = r"D:\aaa"
def generar_isomeros_y_guardar(input_sdf, output_sdf):
"""Genera todos los isómeros de cada molécula en un archivo .sdf y los guarda en un nuevo archivo."""
try:
# Leer moléculas del archivo SDF
supl = Chem.SDMolSupplier(input_sdf, removeHs=False)
mols = [mol for mol in supl if mol is not None]
# Preparar archivo de salida
writer = Chem.SDWriter(output_sdf)
for mol_idx, mol in enumerate(tqdm(mols, desc=f"Procesando {os.path.basename(input_sdf)}", unit="mol")):
# Generar todos los isómeros posibles
options = StereoEnumerationOptions(onlyUnassigned=False)
isomeros = list(EnumerateStereoisomers(mol, options=options))
for iso_idx, isomero in enumerate(isomeros):
# Agregar hidrógenos y generar conformación 3D
isomero = Chem.AddHs(isomero)
AllChem.EmbedMolecule(isomero, randomSeed=42)
# Asignar nombre a cada isómero
isomero.SetProp("_Name", f"{mol.GetProp('_Name')}_{iso_idx + 1}" if mol.HasProp("_Name") else f"Mol_{mol_idx + 1}_{iso_idx + 1}")
# Escribir en el archivo de salida
writer.write(isomero)
writer.close()
except Exception as e:
print(f"Error procesando {input_sdf}: {e}")
def procesar_archivos_sdf(ruta):
"""Procesa todos los archivos .sdf en la ruta dada y genera los archivos de salida con isómeros."""
archivos_sdf = [f for f in os.listdir(ruta) if f.endswith('.sdf')]
if not archivos_sdf:
print("No se encontraron archivos .sdf en la ruta especificada.")
return
for archivo in archivos_sdf:
input_sdf = os.path.join(ruta, archivo)
output_sdf = os.path.join(ruta, f"{os.path.splitext(archivo)[0]}_isomeros.sdf")
print(f"\nGenerando isómeros para {archivo}...")
generar_isomeros_y_guardar(input_sdf, output_sdf)
# Ejecutar la función principal
if __name__ == "__main__":
procesar_archivos_sdf(ruta_sdf)
Una versión equivalente en Linux...
# -*- coding: utf-8 -*-
import os
from rdkit import Chem
from rdkit.Chem import AllChem
from rdkit.Chem.EnumerateStereoisomers import EnumerateStereoisomers, StereoEnumerationOptions
from tqdm import tqdm
# Ruta donde se encuentran los archivos .sdf
ruta_sdf = "/home/jant.encinar/runQuiral/Q12"
def generar_isomeros_y_guardar(input_sdf, output_sdf):
"""Genera todos los isomeros de cada molecula en un archivo .sdf y los guarda en un nuevo archivo."""
try:
# Leer moleculas del archivo SDF
supl = Chem.SDMolSupplier(input_sdf, removeHs=False)
mols = [mol for mol in supl if mol is not None]
# Preparar archivo de salida
writer = Chem.SDWriter(output_sdf)
for mol_idx, mol in enumerate(tqdm(mols, desc=f"Procesando {os.path.basename(input_sdf)}", unit="mol")):
# Generar todos los isomeros posibles
options = StereoEnumerationOptions(onlyUnassigned=False)
isomeros = list(EnumerateStereoisomers(mol, options=options))
for iso_idx, isomero in enumerate(isomeros):
# Agregar hidrogenos y generar conformacion 3D
isomero = Chem.AddHs(isomero)
AllChem.EmbedMolecule(isomero, randomSeed=42)
# Asignar nombre a cada isomero
isomero.SetProp("_Name", f"{mol.GetProp('_Name')}_{iso_idx + 1}" if mol.HasProp("_Name") else f"Mol_{mol_idx + 1}_{iso_idx + 1}")
# Escribir en el archivo de salida
writer.write(isomero)
writer.close()
except Exception as e:
print(f"Error procesando {input_sdf}: {e}")
def procesar_archivos_sdf(ruta):
"""Procesa todos los archivos .sdf en la ruta dada y genera los archivos de salida con isomeros."""
archivos_sdf = [f for f in os.listdir(ruta) if f.endswith('.sdf')]
if not archivos_sdf:
print("No se encontraron archivos .sdf en la ruta especificada.")
return
for archivo in archivos_sdf:
input_sdf = os.path.join(ruta, archivo)
output_sdf = os.path.join(ruta, f"{os.path.splitext(archivo)[0]}_isomeros.sdf")
print(f"\nGenerando isomeros para {archivo}...")
generar_isomeros_y_guardar(input_sdf, output_sdf)
# Ejecutar la funcion principal
if __name__ == "__main__":
procesar_archivos_sdf(ruta_sdf)Una versión diferente en Linux...
# -*- coding: utf-8 -*-
import os
from rdkit import Chem
from rdkit.Chem import AllChem
from rdkit.Chem.EnumerateStereoisomers import EnumerateStereoisomers, StereoEnumerationOptions
from tqdm import tqdm
# Ruta donde se encuentran los archivos .sdf
ruta_sdf = "/home/jant.encinar/runQuiral/Q01"
def generar_isomeros_y_guardar(input_sdf, output_sdf):
"""Genera todos los isomeros de cada molecula en un archivo .sdf y los guarda en un nuevo archivo."""
try:
# Leer moleculas del archivo SDF
supl = Chem.SDMolSupplier(input_sdf, removeHs=False)
mols = [mol for mol in supl if mol is not None]
# Preparar archivo de salida
writer = Chem.SDWriter(output_sdf)
for mol_idx, mol in enumerate(tqdm(mols, desc=f"Procesando {os.path.basename(input_sdf)}", unit="mol")):
# Generar todos los isomeros posibles
options = StereoEnumerationOptions(onlyUnassigned=False)
isomeros = list(EnumerateStereoisomers(mol, options=options))
for iso_idx, isomero in enumerate(isomeros):
# Agregar hidrogenos y generar conformacion 3D
isomero = Chem.AddHs(isomero)
AllChem.EmbedMolecule(isomero, randomSeed=42)
# Asignar nombre a cada isomero
isomero.SetProp("_Name", f"{mol.GetProp('_Name')}_{iso_idx + 1}" if mol.HasProp("_Name") else f"Mol_{mol_idx + 1}_{iso_idx + 1}")
# Escribir en el archivo de salida
writer.write(isomero)
writer.close()
except Exception as e:
print(f"Error procesando {input_sdf}: {e}")
def procesar_archivos_sdf(ruta):
"""Procesa todos los archivos .sdf en la ruta dada y genera los archivos de salida con isomeros."""
archivos_sdf = [f for f in os.listdir(ruta) if f.endswith('.sdf')]
if not archivos_sdf:
print("No se encontraron archivos .sdf en la ruta especificada.")
return
for archivo in archivos_sdf:
input_sdf = os.path.join(ruta, archivo)
output_sdf = os.path.join(ruta, f"{os.path.splitext(archivo)[0]}_isomeros.sdf")
print(f"\nGenerando isomeros para {archivo}...")
generar_isomeros_y_guardar(input_sdf, output_sdf)
# Ejecutar la funcion principal
if __name__ == "__main__":
procesar_archivos_sdf(ruta_sdf)Fichero Numero_de_moleculas
MP7-Q-0001_isomeros_3D.sdf 12696
MP7-Q-0002_isomeros_3D.sdf 7435
MP7-Q-0003_isomeros_3D.sdf 14922
MP7-Q-0004_isomeros_3D.sdf 9597
MP7-Q-0005_isomeros_3D.sdf 18442
MP7-Q-0006_isomeros_3D.sdf 19095
MP7-Q-0007_isomeros_3D.sdf 13696
MP7-Q-0008_isomeros_3D.sdf 8015
MP7-Q-0009_isomeros_3D.sdf 10909
MP7-Q-0010_isomeros_3D.sdf 8912
MP7-Q-0011_isomeros_3D.sdf 28627
MP7-Q-0012_isomeros_3D.sdf 20861
MP7-Q-0013_isomeros_3D.sdf 20326
MP7-Q-0014_isomeros_3D.sdf 11064
MP7-Q-0015_isomeros_3D.sdf 9808
MP7-Q-0016_isomeros_3D.sdf 11729
MP7-Q-0017_isomeros_3D.sdf 13149
MP7-Q-0018_isomeros_3D.sdf 12730
MP7-Q-0019_isomeros_3D.sdf 8423
MP7-Q-0020_isomeros_3D.sdf 10118
MP7-Q-0021_isomeros_3D.sdf 11117
MP7-Q-0022_isomeros_3D.sdf 16672
MP7-Q-0023_isomeros_3D.sdf 8739
MP7-Q-0024_isomeros_3D.sdf 44812
MP7-Q-0025_isomeros_3D.sdf 25745
MP7-Q-0026_isomeros_3D.sdf 22267
MP7-Q-0027_isomeros_3D.sdf 8422
MP7-Q-0028_isomeros_3D.sdf 9010
MP7-Q-0029_isomeros_3D.sdf 8494
MP7-Q-0030_isomeros_3D.sdf 11380
MP7-Q-0031_isomeros_3D.sdf 12774
MP7-Q-0032_isomeros_3D.sdf 8373
MP7-Q-0033_isomeros_3D.sdf 14545
MP7-Q-0034_isomeros_3D.sdf 11466
MP7-Q-0035_isomeros_3D.sdf 9487
MP7-Q-0036_isomeros_3D.sdf 15690
MP7-Q-0037_isomeros_3D.sdf 22650
MP7-Q-0038_isomeros_3D.sdf 7289
MP7-Q-0039_isomeros_3D.sdf 46277
MP7-Q-0040_isomeros_3D.sdf 14920
MP7-Q-0041_isomeros_3D.sdf 20784
MP7-Q-0042_isomeros_3D.sdf 12431
MP7-Q-0043_isomeros_3D.sdf 12774
MP7-Q-0044_isomeros_3D.sdf 9830
MP7-Q-0045_isomeros_3D.sdf 11643
MP7-Q-0046_isomeros_3D.sdf 18055
MP7-Q-0047_isomeros_3D.sdf 10250
MP7-Q-0048_isomeros_3D.sdf 7285
MP7-Q-0049_isomeros_3D.sdf 6264
MP7-Q-0050_isomeros_3D.sdf 6309
MP7-Q-0051_isomeros_3D.sdf 8082
MP7-Q-0052_isomeros_3D.sdf 7355
MP7-Q-0053_isomeros_3D.sdf 6976
MP7-Q-0054_isomeros_3D.sdf 7204
MP7-Q-0055_isomeros_3D.sdf 6665
MP7-Q-0056_isomeros_3D.sdf 7614
MP7-Q-0057_isomeros_3D.sdf 6742
MP7-Q-0058_isomeros_3D.sdf 6574
MP7-Q-0059_isomeros_3D.sdf 25729
MP7-Q-0060_isomeros_3D.sdf 11778
MP7-Q-0061_isomeros_3D.sdf 33835
MP7-Q-0062_isomeros_3D.sdf 7654
MP7-Q-0063_isomeros_3D.sdf 7299
MP7-Q-0064_isomeros_3D.sdf 7065
MP7-Q-0065_isomeros_3D.sdf 7365
MP7-Q-0066_isomeros_3D.sdf 6960
MP7-Q-0067_isomeros_3D.sdf 8258
MP7-Q-0068_isomeros_3D.sdf 7860
MP7-Q-0069_isomeros_3D.sdf 7665
MP7-Q-0070_isomeros_3D.sdf 7849
MP7-Q-0071_isomeros_3D.sdf 7294
MP7-Q-0072_isomeros_3D.sdf 7052
MP7-Q-0073_isomeros_3D.sdf 18764
MP7-Q-0074_isomeros_3D.sdf 9539
MP7-Q-0075_isomeros_3D.sdf 15212
MP7-Q-0076_isomeros_3D.sdf 6897
MP7-Q-0077_isomeros_3D.sdf 7195
MP7-Q-0078_isomeros_3D.sdf 7115
MP7-Q-0079_isomeros_3D.sdf 6986
MP7-Q-0080_isomeros_3D.sdf 7076
MP7-Q-0081_isomeros_3D.sdf 7039
MP7-Q-0082_isomeros_3D.sdf 6963
MP7-Q-0083_isomeros_3D.sdf 7154
MP7-Q-0084_isomeros_3D.sdf 7048
MP7-Q-0085_isomeros_3D.sdf 7082
MP7-Q-0086_isomeros_3D.sdf 7179
MP7-Q-0087_isomeros_3D.sdf 8657
MP7-Q-0088_isomeros_3D.sdf 7969
MP7-Q-0089_isomeros_3D.sdf 9028
MP7-Q-0090_isomeros_3D.sdf 7187
MP7-Q-0091_isomeros_3D.sdf 8480
MP7-Q-0092_isomeros_3D.sdf 6913
MP7-Q-0093_isomeros_3D.sdf 6893
MP7-Q-0094_isomeros_3D.sdf 6971
MP7-Q-0095_isomeros_3D.sdf 7034
MP7-Q-0096_isomeros_3D.sdf 6851
MP7-Q-0097_isomeros_3D.sdf 7134
MP7-Q-0098_isomeros_3D.sdf 7950
MP7-Q-0099_isomeros_3D.sdf 8159
MP7-Q-0100_isomeros_3D.sdf 7460
MP7-Q-0101_isomeros_3D.sdf 7850
MP7-Q-0102_isomeros_3D.sdf 18516
MP7-Q-0103_isomeros_3D.sdf 15458
MP7-Q-0104_isomeros_3D.sdf 33808
MP7-Q-0105_isomeros_3D.sdf 8208
MP7-Q-0106_isomeros_3D.sdf 29308
MP7-Q-0107_isomeros_3D.sdf 7465
MP7-Q-0108_isomeros_3D.sdf 9223
MP7-Q-0109_isomeros_3D.sdf 22440
MP7-Q-0110_isomeros_3D.sdf 6902
MP7-Q-0111_isomeros_3D.sdf 7556
MP7-Q-0112_isomeros_3D.sdf 23686
MP7-Q-0113_isomeros_3D.sdf 13665
MP7-Q-0114_isomeros_3D.sdf 21170
MP7-Q-0115_isomeros_3D.sdf 18774
MP7-Q-0116_isomeros_3D.sdf 8076
MP7-Q-0117_isomeros_3D.sdf 7878
MP7-Q-0118_isomeros_3D.sdf 6909
MP7-Q-0119_isomeros_3D.sdf 7491
MP7-Q-0120_isomeros_3D.sdf 7096
MP7-Q-0121_isomeros_3D.sdf 7039
MP7-Q-0122_isomeros_3D.sdf 7216
MP7-Q-0123_isomeros_3D.sdf 7249
MP7-Q-0124_isomeros_3D.sdf 7044
MP7-Q-0125_isomeros_3D.sdf 7471
MP7-Q-0126_isomeros_3D.sdf 7457
MP7-Q-0127_isomeros_3D.sdf 7241
MP7-Q-0128_isomeros_3D.sdf 7542
MP7-Q-0129_isomeros_3D.sdf 7489
MP7-Q-0130_isomeros_3D.sdf 7140
MP7-Q-0131_isomeros_3D.sdf 19475
MP7-Q-0132_isomeros_3D.sdf 8985
MP7-Q-0133_isomeros_3D.sdf 9439
MP7-Q-0134_isomeros_3D.sdf 8176
MP7-Q-0135_isomeros_3D.sdf 6539
MP7-Q-0136_isomeros_3D.sdf 6208
MP7-Q-0137_isomeros_3D.sdf 8116
MP7-Q-0138_isomeros_3D.sdf 13264
MP7-Q-0139_isomeros_3D.sdf 19805
MP7-Q-0140_isomeros_3D.sdf 8392
MP7-Q-0141_isomeros_3D.sdf 8766
MP7-Q-0142_isomeros_3D.sdf 10674
MP7-Q-0143_isomeros_3D.sdf 10412
MP7-Q-0144_isomeros_3D.sdf 9864
MP7-Q-0145_isomeros_3D.sdf 10735
MP7-Q-0146_isomeros_3D.sdf 6991
MP7-Q-0147_isomeros_3D.sdf 7303
MP7-Q-0148_isomeros_3D.sdf 6993
MP7-Q-0149_isomeros_3D.sdf 8492
MP7-Q-0150_isomeros_3D.sdf 7149
MP7-Q-0151_isomeros_3D.sdf 7100
MP7-Q-0152_isomeros_3D.sdf 8889
MP7-Q-0153_isomeros_3D.sdf 6499
MP7-Q-0154_isomeros_3D.sdf 6852
MP7-Q-0155_isomeros_3D.sdf 7058
MP7-Q-0156_isomeros_3D.sdf 6304
MP7-Q-0157_isomeros_3D.sdf 6531
MP7-Q-0158_isomeros_3D.sdf 6848
MP7-Q-0159_isomeros_3D.sdf 6242
MP7-Q-0160_isomeros_3D.sdf 10002
MP7-Q-0161_isomeros_3D.sdf 7262
MP7-Q-0162_isomeros_3D.sdf 7106
MP7-Q-0163_isomeros_3D.sdf 7701
MP7-Q-0164_isomeros_3D.sdf 10632
MP7-Q-0165_isomeros_3D.sdf 7243
MP7-Q-0166_isomeros_3D.sdf 9404
MP7-Q-0167_isomeros_3D.sdf 7726
MP7-Q-0168_isomeros_3D.sdf 7084
MP7-Q-0169_isomeros_3D.sdf 7889
MP7-Q-0170_isomeros_3D.sdf 3907
MP7-Q-0171_isomeros_3D.sdf 8245
MP7-Q-0172_isomeros_3D.sdf 7084
MP7-Q-0173_isomeros_3D.sdf 7236
MP7-Q-0174_isomeros_3D.sdf 7067
MP7-Q-0175_isomeros_3D.sdf 7210
MP7-Q-0176_isomeros_3D.sdf 7843
MP7-Q-0177_isomeros_3D.sdf 6953
MP7-Q-0178_isomeros_3D.sdf 7368
MP7-Q-0179_isomeros_3D.sdf 7263
MP7-Q-0180_isomeros_3D.sdf 7331
MP7-Q-0181_isomeros_3D.sdf 7757
MP7-Q-0182_isomeros_3D.sdf 7247
MP7-Q-0183_isomeros_3D.sdf 7487
MP7-Q-0184_isomeros_3D.sdf 8541
MP7-Q-0185_isomeros_3D.sdf 14094
MP7-Q-0186_isomeros_3D.sdf 7503
MP7-Q-0187_isomeros_3D.sdf 7419
MP7-Q-0188_isomeros_3D.sdf 7485
MP7-Q-0189_isomeros_3D.sdf 7472
MP7-Q-0190_isomeros_3D.sdf 7575
MP7-Q-0191_isomeros_3D.sdf 7725
MP7-Q-0192_isomeros_3D.sdf 7577
MP7-Q-0193_isomeros_3D.sdf 7356
MP7-Q-0194_isomeros_3D.sdf 7498
MP7-Q-0195_isomeros_3D.sdf 7653
MP7-Q-0196_isomeros_3D.sdf 7718
MP7-Q-0197_isomeros_3D.sdf 7587
MP7-Q-0198_isomeros_3D.sdf 7819
MP7-Q-0199_isomeros_3D.sdf 7718
MP7-Q-0200_isomeros_3D.sdf 8158
MP7-Q-0201_isomeros_3D.sdf 7355
MP7-Q-0202_isomeros_3D.sdf 7840
MP7-Q-0203_isomeros_3D.sdf 8038
MP7-Q-0204_isomeros_3D.sdf 9182
MP7-Q-0205_isomeros_3D.sdf 6736
MP7-Q-0206_isomeros_3D.sdf 6621
MP7-Q-0207_isomeros_3D.sdf 6211
MP7-Q-0208_isomeros_3D.sdf 6137
MP7-Q-0209_isomeros_3D.sdf 6328
MP7-Q-0210_isomeros_3D.sdf 6662
MP7-Q-0211_isomeros_3D.sdf 6993
MP7-Q-0212_isomeros_3D.sdf 7292
MP7-Q-0213_isomeros_3D.sdf 7377
MP7-Q-0214_isomeros_3D.sdf 7525
MP7-Q-0215_isomeros_3D.sdf 7177
MP7-Q-0216_isomeros_3D.sdf 7311
MP7-Q-0217_isomeros_3D.sdf 7071
MP7-Q-0218_isomeros_3D.sdf 7246
MP7-Q-0219_isomeros_3D.sdf 7202
MP7-Q-0220_isomeros_3D.sdf 7704
MP7-Q-0221_isomeros_3D.sdf 7420
MP7-Q-0222_isomeros_3D.sdf 7657
MP7-Q-0223_isomeros_3D.sdf 8494
MP7-Q-0224_isomeros_3D.sdf 8501
MP7-Q-0225_isomeros_3D.sdf 7551
MP7-Q-0226_isomeros_3D.sdf 10709
MP7-Q-0227_isomeros_3D.sdf 6811
MP7-Q-0228_isomeros_3D.sdf 7743
MP7-Q-0229_isomeros_3D.sdf 7059
MP7-Q-0230_isomeros_3D.sdf 7536
MP7-Q-0231_isomeros_3D.sdf 7716
MP7-Q-0232_isomeros_3D.sdf 7520
MP7-Q-0233_isomeros_3D.sdf 7019
MP7-Q-0234_isomeros_3D.sdf 8121
MP7-Q-0235_isomeros_3D.sdf 6887
MP7-Q-0236_isomeros_3D.sdf 8287
MP7-Q-0237_isomeros_3D.sdf 7618
MP7-Q-0238_isomeros_3D.sdf 8044
MP7-Q-0239_isomeros_3D.sdf 7518
MP7-Q-0240_isomeros_3D.sdf 7507
MP7-Q-0241_isomeros_3D.sdf 7464
MP7-Q-0242_isomeros_3D.sdf 6950
MP7-Q-0243_isomeros_3D.sdf 7503
MP7-Q-0244_isomeros_3D.sdf 7937
MP7-Q-0245_isomeros_3D.sdf 7110
MP7-Q-0246_isomeros_3D.sdf 8176
MP7-Q-0247_isomeros_3D.sdf 7288
MP7-Q-0248_isomeros_3D.sdf 7168
MP7-Q-0249_isomeros_3D.sdf 7592
MP7-Q-0250_isomeros_3D.sdf 7343
MP7-Q-0251_isomeros_3D.sdf 6922
MP7-Q-0252_isomeros_3D.sdf 7294
MP7-Q-0253_isomeros_3D.sdf 6725
MP7-Q-0254_isomeros_3D.sdf 7804
MP7-Q-0255_isomeros_3D.sdf 7144
MP7-Q-0256_isomeros_3D.sdf 4087
MP7-Q-0257_isomeros_3D.sdf 8248
MP7-Q-0258_isomeros_3D.sdf 7576
MP7-Q-0259_isomeros_3D.sdf 7410
MP7-Q-0260_isomeros_3D.sdf 7184
MP7-Q-0261_isomeros_3D.sdf 7251
MP7-Q-0262_isomeros_3D.sdf 7318
MP7-Q-0263_isomeros_3D.sdf 7144
MP7-Q-0264_isomeros_3D.sdf 7909
MP7-Q-0265_isomeros_3D.sdf 7325
MP7-Q-0266_isomeros_3D.sdf 7355
MP7-Q-0267_isomeros_3D.sdf 7770
MP7-Q-0268_isomeros_3D.sdf 7595
MP7-Q-0269_isomeros_3D.sdf 7942
MP7-Q-0270_isomeros_3D.sdf 8060
MP7-Q-0271_isomeros_3D.sdf 8190
MP7-Q-0272_isomeros_3D.sdf 7084
MP7-Q-0273_isomeros_3D.sdf 7520
MP7-Q-0274_isomeros_3D.sdf 7288
MP7-Q-0275_isomeros_3D.sdf 7024
MP7-Q-0276_isomeros_3D.sdf 8040
MP7-Q-0277_isomeros_3D.sdf 7107
MP7-Q-0278_isomeros_3D.sdf 7913
MP7-Q-0279_isomeros_3D.sdf 8519
MP7-Q-0280_isomeros_3D.sdf 7686
MP7-Q-0281_isomeros_3D.sdf 7154
MP7-Q-0282_isomeros_3D.sdf 7630
MP7-Q-0283_isomeros_3D.sdf 7868
MP7-Q-0284_isomeros_3D.sdf 7721
MP7-Q-0285_isomeros_3D.sdf 7826
MP7-Q-0286_isomeros_3D.sdf 7588
MP7-Q-0287_isomeros_3D.sdf 7576
MP7-Q-0288_isomeros_3D.sdf 7258
MP7-Q-0289_isomeros_3D.sdf 7185
MP7-Q-0290_isomeros_3D.sdf 7072
MP7-Q-0291_isomeros_3D.sdf 8194
MP7-Q-0292_isomeros_3D.sdf 7485
MP7-Q-0293_isomeros_3D.sdf 16963
MP7-Q-0294_isomeros_3D.sdf 9148
MP7-Q-0295_isomeros_3D.sdf 9317
MP7-Q-0296_isomeros_3D.sdf 13967
MP7-Q-0297_isomeros_3D.sdf 10961
MP7-Q-0298_isomeros_3D.sdf 10141
MP7-Q-0299_isomeros_3D.sdf 10435
MP7-Q-0300_isomeros_3D.sdf 8806
MP7-Q-0301_isomeros_3D.sdf 8558
MP7-Q-0302_isomeros_3D.sdf 9700
MP7-Q-0303_isomeros_3D.sdf 7938
MP7-Q-0304_isomeros_3D.sdf 9201
MP7-Q-0305_isomeros_3D.sdf 8649
MP7-Q-0306_isomeros_3D.sdf 8253
MP7-Q-0307_isomeros_3D.sdf 7714
MP7-Q-0308_isomeros_3D.sdf 6683
MP7-Q-0309_isomeros_3D.sdf 8384
MP7-Q-0310_isomeros_3D.sdf 7378
MP7-Q-0311_isomeros_3D.sdf 7180
MP7-Q-0312_isomeros_3D.sdf 7416
MP7-Q-0313_isomeros_3D.sdf 7440
MP7-Q-0314_isomeros_3D.sdf 7222
MP7-Q-0315_isomeros_3D.sdf 6987
MP7-Q-0316_isomeros_3D.sdf 12295
MP7-Q-0317_isomeros_3D.sdf 8590
MP7-Q-0318_isomeros_3D.sdf 7853
MP7-Q-0319_isomeros_3D.sdf 7177
MP7-Q-0320_isomeros_3D.sdf 8879
MP7-Q-0321_isomeros_3D.sdf 13310
MP7-Q-0322_isomeros_3D.sdf 12370
MP7-Q-0323_isomeros_3D.sdf 19557
MP7-Q-0324_isomeros_3D.sdf 10193
MP7-Q-0325_isomeros_3D.sdf 9996
MP7-Q-0326_isomeros_3D.sdf 11653
MP7-Q-0327_isomeros_3D.sdf 12325
MP7-Q-0328_isomeros_3D.sdf 7921
MP7-Q-0329_isomeros_3D.sdf 13059
MP7-Q-0330_isomeros_3D.sdf 12013
MP7-Q-0331_isomeros_3D.sdf 10064
MP7-Q-0332_isomeros_3D.sdf 8890
MP7-Q-0333_isomeros_3D.sdf 15883
MP7-Q-0334_isomeros_3D.sdf 8643
MP7-Q-0335_isomeros_3D.sdf 12734
MP7-Q-0336_isomeros_3D.sdf 8413
MP7-Q-0337_isomeros_3D.sdf 7542
MP7-Q-0338_isomeros_3D.sdf 8622
MP7-Q-0339_isomeros_3D.sdf 7165
MP7-Q-0340_isomeros_3D.sdf 10924
MP7-Q-0341_isomeros_3D.sdf 9480
MP7-Q-0342_isomeros_3D.sdf 8651
MP7-Q-0343_isomeros_3D.sdf 9577
MP7-Q-0344_isomeros_3D.sdf 16217
MP7-Q-0345_isomeros_3D.sdf 9139
MP7-Q-0346_isomeros_3D.sdf 8735
MP7-Q-0347_isomeros_3D.sdf 9984
MP7-Q-0348_isomeros_3D.sdf 10521
MP7-Q-0349_isomeros_3D.sdf 7582
MP7-Q-0350_isomeros_3D.sdf 9171
MP7-Q-0351_isomeros_3D.sdf 10727
MP7-Q-0352_isomeros_3D.sdf 8864
MP7-Q-0353_isomeros_3D.sdf 7374
MP7-Q-0354_isomeros_3D.sdf 7637
MP7-Q-0355_isomeros_3D.sdf 8158
MP7-Q-0356_isomeros_3D.sdf 14756
MP7-Q-0357_isomeros_3D.sdf 8380
MP7-Q-0358_isomeros_3D.sdf 7870
MP7-Q-0359_isomeros_3D.sdf 7905
MP7-Q-0360_isomeros_3D.sdf 8673
MP7-Q-0361_isomeros_3D.sdf 7931
MP7-Q-0362_isomeros_3D.sdf 8646
MP7-Q-0363_isomeros_3D.sdf 10256
MP7-Q-0364_isomeros_3D.sdf 8516
MP7-Q-0365_isomeros_3D.sdf 28017
MP7-Q-0366_isomeros_3D.sdf 8362
MP7-Q-0367_isomeros_3D.sdf 7608
MP7-Q-0368_isomeros_3D.sdf 8103
MP7-Q-0369_isomeros_3D.sdf 7647
MP7-Q-0370_isomeros_3D.sdf 8176
MP7-Q-0371_isomeros_3D.sdf 7517
MP7-Q-0372_isomeros_3D.sdf 8241
MP7-Q-0373_isomeros_3D.sdf 7976
MP7-Q-0374_isomeros_3D.sdf 7540
MP7-Q-0375_isomeros_3D.sdf 8164
MP7-Q-0376_isomeros_3D.sdf 7874
MP7-Q-0377_isomeros_3D.sdf 7416
MP7-Q-0378_isomeros_3D.sdf 8826
MP7-Q-0379_isomeros_3D.sdf 9898
MP7-Q-0380_isomeros_3D.sdf 10127
MP7-Q-0381_isomeros_3D.sdf 10002
MP7-Q-0382_isomeros_3D.sdf 10141
MP7-Q-0383_isomeros_3D.sdf 10017
MP7-Q-0384_isomeros_3D.sdf 10100
MP7-Q-0385_isomeros_3D.sdf 8908
MP7-Q-0386_isomeros_3D.sdf 6053
MP7-Q-0387_isomeros_3D.sdf 6186
MP7-Q-0388_isomeros_3D.sdf 6002
MP7-Q-0389_isomeros_3D.sdf 6046
MP7-Q-0390_isomeros_3D.sdf 6078
MP7-Q-0391_isomeros_3D.sdf 7973
MP7-Q-0392_isomeros_3D.sdf 8834
MP7-Q-0393_isomeros_3D.sdf 13752
MP7-Q-0394_isomeros_3D.sdf 9860
MP7-Q-0395_isomeros_3D.sdf 7818
MP7-Q-0396_isomeros_3D.sdf 7956
MP7-Q-0397_isomeros_3D.sdf 9749
MP7-Q-0398_isomeros_3D.sdf 8201
MP7-Q-0399_isomeros_3D.sdf 7111
MP7-Q-0400_isomeros_3D.sdf 7217
MP7-Q-0401_isomeros_3D.sdf 9276
MP7-Q-0402_isomeros_3D.sdf 8131
MP7-Q-0403_isomeros_3D.sdf 8126
MP7-Q-0404_isomeros_3D.sdf 7481
MP7-Q-0405_isomeros_3D.sdf 10440
MP7-Q-0406_isomeros_3D.sdf 13239
MP7-Q-0407_isomeros_3D.sdf 10988
MP7-Q-0408_isomeros_3D.sdf 8213
MP7-Q-0409_isomeros_3D.sdf 10872
MP7-Q-0410_isomeros_3D.sdf 8527
MP7-Q-0411_isomeros_3D.sdf 8546
MP7-Q-0412_isomeros_3D.sdf 8558
MP7-Q-0413_isomeros_3D.sdf 10521
MP7-Q-0414_isomeros_3D.sdf 23601
MP7-Q-0415_isomeros_3D.sdf 6432
MP7-Q-0416_isomeros_3D.sdf 8258
MP7-Q-0417_isomeros_3D.sdf 7357
MP7-Q-0418_isomeros_3D.sdf 8096
MP7-Q-0419_isomeros_3D.sdf 7500
MP7-Q-0420_isomeros_3D.sdf 6984
MP7-Q-0421_isomeros_3D.sdf 7678
MP7-Q-0422_isomeros_3D.sdf 8145
MP7-Q-0423_isomeros_3D.sdf 8854
MP7-Q-0424_isomeros_3D.sdf 10067
MP7-Q-0425_isomeros_3D.sdf 8277
MP7-Q-0426_isomeros_3D.sdf 7816
MP7-Q-0427_isomeros_3D.sdf 19423
MP7-Q-0428_isomeros_3D.sdf 8933
MP7-Q-0429_isomeros_3D.sdf 8542
MP7-Q-0430_isomeros_3D.sdf 8512
MP7-Q-0431_isomeros_3D.sdf 8749
MP7-Q-0432_isomeros_3D.sdf 16616
MP7-Q-0433_isomeros_3D.sdf 9625
MP7-Q-0434_isomeros_3D.sdf 7398
MP7-Q-0435_isomeros_3D.sdf 10370
MP7-Q-0436_isomeros_3D.sdf 8254
MP7-Q-0437_isomeros_3D.sdf 8195
MP7-Q-0438_isomeros_3D.sdf 7464
MP7-Q-0439_isomeros_3D.sdf 8365
MP7-Q-0440_isomeros_3D.sdf 7302
MP7-Q-0441_isomeros_3D.sdf 8006
MP7-Q-0442_isomeros_3D.sdf 7889
MP7-Q-0443_isomeros_3D.sdf 8593
MP7-Q-0444_isomeros_3D.sdf 10782
MP7-Q-0445_isomeros_3D.sdf 8250
MP7-Q-0446_isomeros_3D.sdf 8950
MP7-Q-0447_isomeros_3D.sdf 9161
MP7-Q-0448_isomeros_3D.sdf 7382
MP7-Q-0449_isomeros_3D.sdf 10406
MP7-Q-0450_isomeros_3D.sdf 7511
MP7-Q-0451_isomeros_3D.sdf 7772
MP7-Q-0452_isomeros_3D.sdf 8429
MP7-Q-0453_isomeros_3D.sdf 6902
MP7-Q-0454_isomeros_3D.sdf 16821
MP7-Q-0455_isomeros_3D.sdf 14102
MP7-Q-0456_isomeros_3D.sdf 6952
MP7-Q-0457_isomeros_3D.sdf 8559
MP7-Q-0458_isomeros_3D.sdf 8576
MP7-Q-0459_isomeros_3D.sdf 7601
MP7-Q-0460_isomeros_3D.sdf 9721
MP7-Q-0461_isomeros_3D.sdf 9285
MP7-Q-0462_isomeros_3D.sdf 8355
MP7-Q-0463_isomeros_3D.sdf 7683
MP7-Q-0464_isomeros_3D.sdf 10067
MP7-Q-0465_isomeros_3D.sdf 9903
MP7-Q-0466_isomeros_3D.sdf 9832
MP7-Q-0467_isomeros_3D.sdf 10231
MP7-Q-0468_isomeros_3D.sdf 7921
MP7-Q-0469_isomeros_3D.sdf 7573
MP7-Q-0470_isomeros_3D.sdf 11127
MP7-Q-0471_isomeros_3D.sdf 10266
MP7-Q-0472_isomeros_3D.sdf 9292
MP7-Q-0473_isomeros_3D.sdf 12041
MP7-Q-0474_isomeros_3D.sdf 8068
MP7-Q-0475_isomeros_3D.sdf 8376
MP7-Q-0476_isomeros_3D.sdf 10736
MP7-Q-0477_isomeros_3D.sdf 9050
MP7-Q-0478_isomeros_3D.sdf 9744
MP7-Q-0479_isomeros_3D.sdf 7541
MP7-Q-0480_isomeros_3D.sdf 6923
MP7-Q-0481_isomeros_3D.sdf 7499
MP7-Q-0482_isomeros_3D.sdf 8440
MP7-Q-0483_isomeros_3D.sdf 7820
MP7-Q-0484_isomeros_3D.sdf 7395
MP7-Q-0485_isomeros_3D.sdf 7568
MP7-Q-0486_isomeros_3D.sdf 7431
MP7-Q-0487_isomeros_3D.sdf 7064
MP7-Q-0488_isomeros_3D.sdf 7670
MP7-Q-0489_isomeros_3D.sdf 7250
MP7-Q-0490_isomeros_3D.sdf 9999
MP7-Q-0491_isomeros_3D.sdf 9870
MP7-Q-0492_isomeros_3D.sdf 8023
MP7-Q-0493_isomeros_3D.sdf 8977
MP7-Q-0494_isomeros_3D.sdf 8577
MP7-Q-0495_isomeros_3D.sdf 10256
MP7-Q-0496_isomeros_3D.sdf 8987
MP7-Q-0497_isomeros_3D.sdf 8868
MP7-Q-0498_isomeros_3D.sdf 9276
MP7-Q-0499_isomeros_3D.sdf 17713
MP7-Q-0500_isomeros_3D.sdf 8136
MP7-Q-0501_isomeros_3D.sdf 8590
MP7-Q-0502_isomeros_3D.sdf 8625
MP7-Q-0503_isomeros_3D.sdf 8737
MP7-Q-0504_isomeros_3D.sdf 16795
MP7-Q-0505_isomeros_3D.sdf 19883
MP7-Q-0506_isomeros_3D.sdf 8360
MP7-Q-0507_isomeros_3D.sdf 8346
MP7-Q-0508_isomeros_3D.sdf 8365
MP7-Q-0509_isomeros_3D.sdf 8295
MP7-Q-0510_isomeros_3D.sdf 8174
MP7-Q-0511_isomeros_3D.sdf 9144
MP7-Q-0512_isomeros_3D.sdf 9245
MP7-Q-0513_isomeros_3D.sdf 8764
MP7-Q-0514_isomeros_3D.sdf 12975
MP7-Q-0515_isomeros_3D.sdf 7727
MP7-Q-0516_isomeros_3D.sdf 7738
MP7-Q-0517_isomeros_3D.sdf 7667
MP7-Q-0518_isomeros_3D.sdf 7946
MP7-Q-0519_isomeros_3D.sdf 7759
MP7-Q-0520_isomeros_3D.sdf 7247
MP7-Q-0521_isomeros_3D.sdf 7436
MP7-Q-0522_isomeros_3D.sdf 7668
MP7-Q-0523_isomeros_3D.sdf 7925
MP7-Q-0524_isomeros_3D.sdf 8491
MP7-Q-0525_isomeros_3D.sdf 8359
MP7-Q-0526_isomeros_3D.sdf 8712
MP7-Q-0527_isomeros_3D.sdf 8480
MP7-Q-0528_isomeros_3D.sdf 7863
MP7-Q-0529_isomeros_3D.sdf 7407
MP7-Q-0530_isomeros_3D.sdf 7217
MP7-Q-0531_isomeros_3D.sdf 7529
MP7-Q-0532_isomeros_3D.sdf 7727
MP7-Q-0533_isomeros_3D.sdf 7887
MP7-Q-0534_isomeros_3D.sdf 8428
MP7-Q-0535_isomeros_3D.sdf 8318
MP7-Q-0536_isomeros_3D.sdf 8275
MP7-Q-0537_isomeros_3D.sdf 9031
MP7-Q-0538_isomeros_3D.sdf 14085
MP7-Q-0539_isomeros_3D.sdf 6236
MP7-Q-0540_isomeros_3D.sdf 7725
MP7-Q-0541_isomeros_3D.sdf 7472
MP7-Q-0542_isomeros_3D.sdf 7468
MP7-Q-0543_isomeros_3D.sdf 15218
MP7-Q-0544_isomeros_3D.sdf 8909
MP7-Q-0545_isomeros_3D.sdf 9212
MP7-Q-0546_isomeros_3D.sdf 8107
MP7-Q-0547_isomeros_3D.sdf 8237
MP7-Q-0548_isomeros_3D.sdf 8810
MP7-Q-0549_isomeros_3D.sdf 8546
MP7-Q-0550_isomeros_3D.sdf 9092
MP7-Q-0551_isomeros_3D.sdf 9001
MP7-Q-0552_isomeros_3D.sdf 10508
MP7-Q-0553_isomeros_3D.sdf 6984
MP7-Q-0554_isomeros_3D.sdf 8064
MP7-Q-0555_isomeros_3D.sdf 8004
MP7-Q-0556_isomeros_3D.sdf 12044
MP7-Q-0557_isomeros_3D.sdf 9776
MP7-Q-0558_isomeros_3D.sdf 8994
MP7-Q-0559_isomeros_3D.sdf 8963
MP7-Q-0560_isomeros_3D.sdf 8966
MP7-Q-0561_isomeros_3D.sdf 13102
MP7-Q-0562_isomeros_3D.sdf 8948
MP7-Q-0563_isomeros_3D.sdf 8649
MP7-Q-0564_isomeros_3D.sdf 9700
MP7-Q-0565_isomeros_3D.sdf 6829
MP7-Q-0566_isomeros_3D.sdf 9569
MP7-Q-0567_isomeros_3D.sdf 8323
MP7-Q-0568_isomeros_3D.sdf 7430
MP7-Q-0569_isomeros_3D.sdf 8156
MP7-Q-0570_isomeros_3D.sdf 11210
MP7-Q-0571_isomeros_3D.sdf 8365
MP7-Q-0572_isomeros_3D.sdf 8388
MP7-Q-0573_isomeros_3D.sdf 7573
MP7-Q-0574_isomeros_3D.sdf 9762
MP7-Q-0575_isomeros_3D.sdf 7211
MP7-Q-0576_isomeros_3D.sdf 9436
MP7-Q-0577_isomeros_3D.sdf 9297
MP7-Q-0578_isomeros_3D.sdf 8345
MP7-Q-0579_isomeros_3D.sdf 7287
MP7-Q-0580_isomeros_3D.sdf 7046
MP7-Q-0581_isomeros_3D.sdf 7773
MP7-Q-0582_isomeros_3D.sdf 7429
MP7-Q-0583_isomeros_3D.sdf 7605
MP7-Q-0584_isomeros_3D.sdf 10205
MP7-Q-0585_isomeros_3D.sdf 10986
MP7-Q-0586_isomeros_3D.sdf 9192
MP7-Q-0587_isomeros_3D.sdf 9221
MP7-Q-0588_isomeros_3D.sdf 9362
MP7-Q-0589_isomeros_3D.sdf 8828
MP7-Q-0590_isomeros_3D.sdf 7829
MP7-Q-0591_isomeros_3D.sdf 9266
MP7-Q-0592_isomeros_3D.sdf 12908
MP7-Q-0593_isomeros_3D.sdf 7232
MP7-Q-0594_isomeros_3D.sdf 7712
MP7-Q-0595_isomeros_3D.sdf 9257
MP7-Q-0596_isomeros_3D.sdf 9577
MP7-Q-0597_isomeros_3D.sdf 8293
MP7-Q-0598_isomeros_3D.sdf 8951
MP7-Q-0599_isomeros_3D.sdf 7837
MP7-Q-0600_isomeros_3D.sdf 8367
MP7-Q-0601_isomeros_3D.sdf 11446
MP7-Q-0602_isomeros_3D.sdf 8520
MP7-Q-0603_isomeros_3D.sdf 8310
MP7-Q-0604_isomeros_3D.sdf 9010
MP7-Q-0605_isomeros_3D.sdf 10451
MP7-Q-0606_isomeros_3D.sdf 8224
MP7-Q-0607_isomeros_3D.sdf 9630
MP7-Q-0608_isomeros_3D.sdf 9336
MP7-Q-0609_isomeros_3D.sdf 10129
MP7-Q-0610_isomeros_3D.sdf 7111
MP7-Q-0611_isomeros_3D.sdf 9045
MP7-Q-0612_isomeros_3D.sdf 8098
MP7-Q-0613_isomeros_3D.sdf 10890
MP7-Q-0614_isomeros_3D.sdf 13214
MP7-Q-0615_isomeros_3D.sdf 11460
MP7-Q-0616_isomeros_3D.sdf 11142
MP7-Q-0617_isomeros_3D.sdf 7471
MP7-Q-0618_isomeros_3D.sdf 7145
MP7-Q-0619_isomeros_3D.sdf 10497
MP7-Q-0620_isomeros_3D.sdf 9406
MP7-Q-0621_isomeros_3D.sdf 7330
MP7-Q-0622_isomeros_3D.sdf 8315
MP7-Q-0623_isomeros_3D.sdf 7672
MP7-Q-0624_isomeros_3D.sdf 7518
MP7-Q-0625_isomeros_3D.sdf 8534
MP7-Q-0626_isomeros_3D.sdf 8264
MP7-Q-0627_isomeros_3D.sdf 8135
MP7-Q-0628_isomeros_3D.sdf 9185
MP7-Q-0629_isomeros_3D.sdf 8446
MP7-Q-0630_isomeros_3D.sdf 10408
MP7-Q-0631_isomeros_3D.sdf 9217
MP7-Q-0632_isomeros_3D.sdf 9371
MP7-Q-0633_isomeros_3D.sdf 8769
MP7-Q-0634_isomeros_3D.sdf 9145
MP7-Q-0635_isomeros_3D.sdf 11774
MP7-Q-0636_isomeros_3D.sdf 9080
MP7-Q-0637_isomeros_3D.sdf 8517
MP7-Q-0638_isomeros_3D.sdf 8639
MP7-Q-0639_isomeros_3D.sdf 11424
MP7-Q-0640_isomeros_3D.sdf 7687
MP7-Q-0641_isomeros_3D.sdf 6737
MP7-Q-0642_isomeros_3D.sdf 11627
MP7-Q-0643_isomeros_3D.sdf 7686
MP7-Q-0644_isomeros_3D.sdf 7873
MP7-Q-0645_isomeros_3D.sdf 7507
MP7-Q-0646_isomeros_3D.sdf 7542
MP7-Q-0647_isomeros_3D.sdf 8139
MP7-Q-0648_isomeros_3D.sdf 8403
MP7-Q-0649_isomeros_3D.sdf 7442
MP7-Q-0650_isomeros_3D.sdf 7386
MP7-Q-0651_isomeros_3D.sdf 7287
MP7-Q-0652_isomeros_3D.sdf 13683
MP7-Q-0653_isomeros_3D.sdf 6577
MP7-Q-0654_isomeros_3D.sdf 8538
MP7-Q-0655_isomeros_3D.sdf 9872
MP7-Q-0656_isomeros_3D.sdf 10290
MP7-Q-0657_isomeros_3D.sdf 8100
MP7-Q-0658_isomeros_3D.sdf 9362
MP7-Q-0659_isomeros_3D.sdf 7201
MP7-Q-0660_isomeros_3D.sdf 13213
MP7-Q-0661_isomeros_3D.sdf 5017
MP7-Q-0662_isomeros_3D.sdf 6839
MP7-Q-0663_isomeros_3D.sdf 7368
MP7-Q-0664_isomeros_3D.sdf 6841
MP7-Q-0665_isomeros_3D.sdf 6703
MP7-Q-0666_isomeros_3D.sdf 7240
MP7-Q-0667_isomeros_3D.sdf 9604
MP7-Q-0668_isomeros_3D.sdf 13048
MP7-Q-0669_isomeros_3D.sdf 7622
MP7-Q-0670_isomeros_3D.sdf 8243
MP7-Q-0671_isomeros_3D.sdf 7954
MP7-Q-0672_isomeros_3D.sdf 7561
MP7-Q-0673_isomeros_3D.sdf 7346
MP7-Q-0674_isomeros_3D.sdf 6481
MP7-Q-0675_isomeros_3D.sdf 6669
MP7-Q-0676_isomeros_3D.sdf 7094
MP7-Q-0677_isomeros_3D.sdf 6843
MP7-Q-0678_isomeros_3D.sdf 7120
MP7-Q-0679_isomeros_3D.sdf 7016
MP7-Q-0680_isomeros_3D.sdf 7145
MP7-Q-0681_isomeros_3D.sdf 6548
MP7-Q-0682_isomeros_3D.sdf 7166
MP7-Q-0683_isomeros_3D.sdf 6836
MP7-Q-0684_isomeros_3D.sdf 6506
MP7-Q-0685_isomeros_3D.sdf 6400
MP7-Q-0686_isomeros_3D.sdf 6669
MP7-Q-0687_isomeros_3D.sdf 7504
MP7-Q-0688_isomeros_3D.sdf 8002
MP7-Q-0689_isomeros_3D.sdf 8184
MP7-Q-0690_isomeros_3D.sdf 11682
MP7-Q-0691_isomeros_3D.sdf 18809
MP7-Q-0692_isomeros_3D.sdf 14238
MP7-Q-0693_isomeros_3D.sdf 14150
MP7-Q-0694_isomeros_3D.sdf 13983
MP7-Q-0695_isomeros_3D.sdf 13706
MP7-Q-0696_isomeros_3D.sdf 13397
MP7-Q-0697_isomeros_3D.sdf 7355
MP7-Q-0698_isomeros_3D.sdf 7353
MP7-Q-0699_isomeros_3D.sdf 10223
MP7-Q-0700_isomeros_3D.sdf 7560
MP7-Q-0701_isomeros_3D.sdf 8779
MP7-Q-0702_isomeros_3D.sdf 9543
MP7-Q-0703_isomeros_3D.sdf 9365
MP7-Q-0704_isomeros_3D.sdf 9030
MP7-Q-0705_isomeros_3D.sdf 9370
MP7-Q-0706_isomeros_3D.sdf 7592
MP7-Q-0707_isomeros_3D.sdf 9555
MP7-Q-0708_isomeros_3D.sdf 8569
MP7-Q-0709_isomeros_3D.sdf 9009
MP7-Q-0710_isomeros_3D.sdf 9416
MP7-Q-0711_isomeros_3D.sdf 9222
MP7-Q-0712_isomeros_3D.sdf 7435
MP7-Q-0713_isomeros_3D.sdf 7363
MP7-Q-0714_isomeros_3D.sdf 19364
MP7-Q-0715_isomeros_3D.sdf 8446
MP7-Q-0716_isomeros_3D.sdf 9760
MP7-Q-0717_isomeros_3D.sdf 30979
MP7-Q-0718_isomeros_3D.sdf 9757
MP7-Q-0719_isomeros_3D.sdf 16386
MP7-Q-0720_isomeros_3D.sdf 11302
MP7-Q-0721_isomeros_3D.sdf 10812
MP7-Q-0722_isomeros_3D.sdf 11094
MP7-Q-0723_isomeros_3D.sdf 10194
MP7-Q-0724_isomeros_3D.sdf 10688
MP7-Q-0725_isomeros_3D.sdf 12552
MP7-Q-0726_isomeros_3D.sdf 11700
MP7-Q-0727_isomeros_3D.sdf 10826
MP7-Q-0728_isomeros_3D.sdf 10390
MP7-Q-0729_isomeros_3D.sdf 9983
MP7-Q-0730_isomeros_3D.sdf 10309
MP7-Q-0731_isomeros_3D.sdf 9703
MP7-Q-0732_isomeros_3D.sdf 11402
MP7-Q-0733_isomeros_3D.sdf 10897
MP7-Q-0734_isomeros_3D.sdf 9206
MP7-Q-0735_isomeros_3D.sdf 9302
MP7-Q-0736_isomeros_3D.sdf 10169
MP7-Q-0737_isomeros_3D.sdf 9033
MP7-Q-0738_isomeros_3D.sdf 9762
MP7-Q-0739_isomeros_3D.sdf 8824
MP7-Q-0740_isomeros_3D.sdf 9178
MP7-Q-0741_isomeros_3D.sdf 12680
MP7-Q-0742_isomeros_3D.sdf 11852
MP7-Q-0743_isomeros_3D.sdf 12683
MP7-Q-0744_isomeros_3D.sdf 11351
MP7-Q-0745_isomeros_3D.sdf 15198
MP7-Q-0746_isomeros_3D.sdf 15325
MP7-Q-0747_isomeros_3D.sdf 12792
MP7-Q-0748_isomeros_3D.sdf 12435
MP7-Q-0749_isomeros_3D.sdf 10487
MP7-Q-0750_isomeros_3D.sdf 11037
MP7-Q-0751_isomeros_3D.sdf 12405
MP7-Q-0752_isomeros_3D.sdf 11782
MP7-Q-0753_isomeros_3D.sdf 13785
MP7-Q-0754_isomeros_3D.sdf 13144
MP7-Q-0755_isomeros_3D.sdf 10797
MP7-Q-0756_isomeros_3D.sdf 13255
MP7-Q-0757_isomeros_3D.sdf 12132
MP7-Q-0758_isomeros_3D.sdf 11140
MP7-Q-0759_isomeros_3D.sdf 16307
MP7-Q-0760_isomeros_3D.sdf 16915
MP7-Q-0761_isomeros_3D.sdf 11462
MP7-Q-0762_isomeros_3D.sdf 11422
MP7-Q-0763_isomeros_3D.sdf 11951
MP7-Q-0764_isomeros_3D.sdf 10681
MP7-Q-0765_isomeros_3D.sdf 10459
MP7-Q-0766_isomeros_3D.sdf 12399
MP7-Q-0767_isomeros_3D.sdf 12983
MP7-Q-0768_isomeros_3D.sdf 11072
MP7-Q-0769_isomeros_3D.sdf 15782
MP7-Q-0770_isomeros_3D.sdf 12469
MP7-Q-0771_isomeros_3D.sdf 10884
MP7-Q-0772_isomeros_3D.sdf 13270
MP7-Q-0773_isomeros_3D.sdf 4501
MP7-Q-0774_isomeros_3D.sdf 12979
MP7-Q-0775_isomeros_3D.sdf 15312
MP7-Q-0776_isomeros_3D.sdf 14553
MP7-Q-0777_isomeros_3D.sdf 14311
MP7-Q-0778_isomeros_3D.sdf 14042
MP7-Q-0779_isomeros_3D.sdf 15056
MP7-Q-0780_isomeros_3D.sdf 14504
MP7-Q-0781_isomeros_3D.sdf 16703
MP7-Q-0782_isomeros_3D.sdf 16126
MP7-Q-0783_isomeros_3D.sdf 15128
MP7-Q-0784_isomeros_3D.sdf 13280
MP7-Q-0785_isomeros_3D.sdf 12011
MP7-Q-0786_isomeros_3D.sdf 12333
MP7-Q-0787_isomeros_3D.sdf 12724
MP7-Q-0788_isomeros_3D.sdf 13567
MP7-Q-0789_isomeros_3D.sdf 13779
MP7-Q-0790_isomeros_3D.sdf 13671
MP7-Q-0791_isomeros_3D.sdf 12786
MP7-Q-0792_isomeros_3D.sdf 11717
MP7-Q-0793_isomeros_3D.sdf 12286
MP7-Q-0794_isomeros_3D.sdf 7292
MP7-Q-0795_isomeros_3D.sdf 16354
MP7-Q-0796_isomeros_3D.sdf 16419
MP7-Q-0797_isomeros_3D.sdf 15046
MP7-Q-0798_isomeros_3D.sdf 14915
MP7-Q-0799_isomeros_3D.sdf 13025
MP7-Q-0800_isomeros_3D.sdf 12341
MP7-Q-0801_isomeros_3D.sdf 14639
MP7-Q-0802_isomeros_3D.sdf 13949
MP7-Q-0803_isomeros_3D.sdf 13635
MP7-Q-0804_isomeros_3D.sdf 14342
MP7-Q-0805_isomeros_3D.sdf 12853
MP7-Q-0806_isomeros_3D.sdf 10938
MP7-Q-0807_isomeros_3D.sdf 12444
MP7-Q-0808_isomeros_3D.sdf 13140
MP7-Q-0809_isomeros_3D.sdf 15953
MP7-Q-0810_isomeros_3D.sdf 16411
MP7-Q-0811_isomeros_3D.sdf 19095
MP7-Q-0812_isomeros_3D.sdf 14308
MP7-Q-0813_isomeros_3D.sdf 20129
MP7-Q-0814_isomeros_3D.sdf 15260
MP7-Q-0815_isomeros_3D.sdf 15783
MP7-Q-0816_isomeros_3D.sdf 15744
MP7-Q-0817_isomeros_3D.sdf 17375
MP7-Q-0818_isomeros_3D.sdf 13185
MP7-Q-0819_isomeros_3D.sdf 13557
MP7-Q-0820_isomeros_3D.sdf 13864
MP7-Q-0821_isomeros_3D.sdf 13298
MP7-Q-0822_isomeros_3D.sdf 12196
MP7-Q-0823_isomeros_3D.sdf 14541
MP7-Q-0824_isomeros_3D.sdf 14307
MP7-Q-0825_isomeros_3D.sdf 13638
MP7-Q-0826_isomeros_3D.sdf 12870
MP7-Q-0827_isomeros_3D.sdf 13787
MP7-Q-0828_isomeros_3D.sdf 14263
MP7-Q-0829_isomeros_3D.sdf 14189
MP7-Q-0830_isomeros_3D.sdf 13398
MP7-Q-0831_isomeros_3D.sdf 14189
MP7-Q-0832_isomeros_3D.sdf 4651
MP7-Q-0833_isomeros_3D.sdf 16571
MP7-Q-0834_isomeros_3D.sdf 16803
MP7-Q-0835_isomeros_3D.sdf 16532
MP7-Q-0836_isomeros_3D.sdf 18326
MP7-Q-0837_isomeros_3D.sdf 16910
MP7-Q-0838_isomeros_3D.sdf 16753
MP7-Q-0839_isomeros_3D.sdf 13925
MP7-Q-0840_isomeros_3D.sdf 12713
MP7-Q-0841_isomeros_3D.sdf 10796
MP7-Q-0842_isomeros_3D.sdf 10562
MP7-Q-0843_isomeros_3D.sdf 12835
MP7-Q-0844_isomeros_3D.sdf 12249
MP7-Q-0845_isomeros_3D.sdf 10535
MP7-Q-0846_isomeros_3D.sdf 13140
MP7-Q-0847_isomeros_3D.sdf 14048
MP7-Q-0848_isomeros_3D.sdf 13467
MP7-Q-0849_isomeros_3D.sdf 13340
MP7-Q-0850_isomeros_3D.sdf 13569
MP7-Q-0851_isomeros_3D.sdf 13905
MP7-Q-0852_isomeros_3D.sdf 13927
MP7-Q-0853_isomeros_3D.sdf 14193
MP7-Q-0854_isomeros_3D.sdf 15082
MP7-Q-0855_isomeros_3D.sdf 13226
MP7-Q-0856_isomeros_3D.sdf 14327
MP7-Q-0857_isomeros_3D.sdf 11919
MP7-Q-0858_isomeros_3D.sdf 12230
MP7-Q-0859_isomeros_3D.sdf 11316
MP7-Q-0860_isomeros_3D.sdf 14581
MP7-Q-0861_isomeros_3D.sdf 15119
MP7-Q-0862_isomeros_3D.sdf 14117
MP7-Q-0863_isomeros_3D.sdf 14202
MP7-Q-0864_isomeros_3D.sdf 20748
MP7-Q-0865_isomeros_3D.sdf 18704
MP7-Q-0866_isomeros_3D.sdf 14668
MP7-Q-0867_isomeros_3D.sdf 13203
MP7-Q-0868_isomeros_3D.sdf 10896
MP7-Q-0869_isomeros_3D.sdf 11502
MP7-Q-0870_isomeros_3D.sdf 13357
MP7-Q-0871_isomeros_3D.sdf 14723
MP7-Q-0872_isomeros_3D.sdf 13892
MP7-Q-0873_isomeros_3D.sdf 14277
MP7-Q-0874_isomeros_3D.sdf 13828
MP7-Q-0875_isomeros_3D.sdf 17241
MP7-Q-0876_isomeros_3D.sdf 16520
MP7-Q-0877_isomeros_3D.sdf 17570
MP7-Q-0878_isomeros_3D.sdf 14506
MP7-Q-0879_isomeros_3D.sdf 15706
MP7-Q-0880_isomeros_3D.sdf 18233
MP7-Q-0881_isomeros_3D.sdf 18333
MP7-Q-0882_isomeros_3D.sdf 19805
MP7-Q-0883_isomeros_3D.sdf 20253
MP7-Q-0884_isomeros_3D.sdf 21142
MP7-Q-0885_isomeros_3D.sdf 14575
MP7-Q-0886_isomeros_3D.sdf 15334
MP7-Q-0887_isomeros_3D.sdf 17642
MP7-Q-0888_isomeros_3D.sdf 11922
MP7-Q-0889_isomeros_3D.sdf 9944
MP7-Q-0890_isomeros_3D.sdf 12751
MP7-Q-0891_isomeros_3D.sdf 11329
MP7-Q-0892_isomeros_3D.sdf 12718
MP7-Q-0893_isomeros_3D.sdf 11804
MP7-Q-0894_isomeros_3D.sdf 14933
MP7-Q-0895_isomeros_3D.sdf 14095
MP7-Q-0896_isomeros_3D.sdf 10860
MP7-Q-0897_isomeros_3D.sdf 11132
MP7-Q-0898_isomeros_3D.sdf 10111
MP7-Q-0899_isomeros_3D.sdf 11147
MP7-Q-0900_isomeros_3D.sdf 14005
MP7-Q-0901_isomeros_3D.sdf 15068
MP7-Q-0902_isomeros_3D.sdf 13138
MP7-Q-0903_isomeros_3D.sdf 14550
MP7-Q-0904_isomeros_3D.sdf 14182
MP7-Q-0905_isomeros_3D.sdf 15978
MP7-Q-0906_isomeros_3D.sdf 16321
MP7-Q-0907_isomeros_3D.sdf 15240
MP7-Q-0908_isomeros_3D.sdf 13930
MP7-Q-0909_isomeros_3D.sdf 13250
MP7-Q-0910_isomeros_3D.sdf 12998
MP7-Q-0911_isomeros_3D.sdf 12139
MP7-Q-0912_isomeros_3D.sdf 11944
MP7-Q-0913_isomeros_3D.sdf 10823
MP7-Q-0914_isomeros_3D.sdf 11039
MP7-Q-0915_isomeros_3D.sdf 11610
MP7-Q-0916_isomeros_3D.sdf 12742
MP7-Q-0917_isomeros_3D.sdf 13438
MP7-Q-0918_isomeros_3D.sdf 13129
MP7-Q-0919_isomeros_3D.sdf 11976
MP7-Q-0920_isomeros_3D.sdf 12374
MP7-Q-0921_isomeros_3D.sdf 11806
MP7-Q-0922_isomeros_3D.sdf 13373
MP7-Q-0923_isomeros_3D.sdf 14698
MP7-Q-0924_isomeros_3D.sdf 13959
MP7-Q-0925_isomeros_3D.sdf 12941
MP7-Q-0926_isomeros_3D.sdf 13676
MP7-Q-0927_isomeros_3D.sdf 12917
MP7-Q-0928_isomeros_3D.sdf 12538
MP7-Q-0929_isomeros_3D.sdf 16489
MP7-Q-0930_isomeros_3D.sdf 14560
MP7-Q-0931_isomeros_3D.sdf 12050
MP7-Q-0932_isomeros_3D.sdf 13854
MP7-Q-0933_isomeros_3D.sdf 13842
MP7-Q-0934_isomeros_3D.sdf 14446
MP7-Q-0935_isomeros_3D.sdf 13410
MP7-Q-0936_isomeros_3D.sdf 13948
MP7-Q-0937_isomeros_3D.sdf 5289
MP7-Q-0938_isomeros_3D.sdf 408
MP7-Q-0939_isomeros_3D.sdf 2421
MP7-Q-0940_isomeros_3D.sdf 12332
MP7-Q-0941_isomeros_3D.sdf 13318
MP7-Q-0942_isomeros_3D.sdf 19143
MP7-Q-0943_isomeros_3D.sdf 21382
MP7-Q-0944_isomeros_3D.sdf 14583
MP7-Q-0945_isomeros_3D.sdf 15358
MP7-Q-0946_isomeros_3D.sdf 13527
MP7-Q-0947_isomeros_3D.sdf 12758
MP7-Q-0948_isomeros_3D.sdf 15451
MP7-Q-0949_isomeros_3D.sdf 14824
MP7-Q-0950_isomeros_3D.sdf 14705
MP7-Q-0951_isomeros_3D.sdf 14051
MP7-Q-0952_isomeros_3D.sdf 11638
MP7-Q-0953_isomeros_3D.sdf 18022
MP7-Q-0954_isomeros_3D.sdf 17702
MP7-Q-0955_isomeros_3D.sdf 16518
MP7-Q-0956_isomeros_3D.sdf 17988
MP7-Q-0957_isomeros_3D.sdf 20791
MP7-Q-0958_isomeros_3D.sdf 16496
MP7-Q-0959_isomeros_3D.sdf 15020
MP7-Q-0960_isomeros_3D.sdf 19167
MP7-Q-0961_isomeros_3D.sdf 20818
MP7-Q-0962_isomeros_3D.sdf 13896
MP7-Q-0963_isomeros_3D.sdf 14471
MP7-Q-0964_isomeros_3D.sdf 11556
MP7-Q-0965_isomeros_3D.sdf 13367
MP7-Q-0966_isomeros_3D.sdf 14581
MP7-Q-0967_isomeros_3D.sdf 10945
MP7-Q-0968_isomeros_3D.sdf 13216
MP7-Q-0969_isomeros_3D.sdf 14172
MP7-Q-0970_isomeros_3D.sdf 20103
MP7-Q-0971_isomeros_3D.sdf 21644
MP7-Q-0972_isomeros_3D.sdf 21592
MP7-Q-0973_isomeros_3D.sdf 20750
MP7-Q-0974_isomeros_3D.sdf 16758
MP7-Q-0975_isomeros_3D.sdf 12762
MP7-Q-0976_isomeros_3D.sdf 14910
MP7-Q-0977_isomeros_3D.sdf 16207
MP7-Q-0978_isomeros_3D.sdf 640
MP7-Q-0979_isomeros_3D.sdf 11548
MP7-Q-0980_isomeros_3D.sdf 14138
MP7-Q-0981_isomeros_3D.sdf 16447
MP7-Q-0982_isomeros_3D.sdf 14480
MP7-Q-0983_isomeros_3D.sdf 13636
MP7-Q-0984_isomeros_3D.sdf 14940
MP7-Q-0985_isomeros_3D.sdf 17463
MP7-Q-0986_isomeros_3D.sdf 18215
MP7-Q-0987_isomeros_3D.sdf 14314
MP7-Q-0988_isomeros_3D.sdf 13751
MP7-Q-0989_isomeros_3D.sdf 14750
MP7-Q-0990_isomeros_3D.sdf 16330
MP7-Q-0991_isomeros_3D.sdf 12022
MP7-Q-0992_isomeros_3D.sdf 9245
MP7-Q-0993_isomeros_3D.sdf 8265
MP7-Q-0994_isomeros_3D.sdf 8176
MP7-Q-0995_isomeros_3D.sdf 8687
MP7-Q-0996_isomeros_3D.sdf 8816
MP7-Q-0997_isomeros_3D.sdf 8295
MP7-Q-0998_isomeros_3D.sdf 9130
MP7-Q-0999_isomeros_3D.sdf 9360
MP7-Q-1000_isomeros_3D.sdf 8209
MP7-Q-1001_isomeros_3D.sdf 9692
MP7-Q-1002_isomeros_3D.sdf 12477
MP7-Q-1003_isomeros_3D.sdf 9690
MP7-Q-1004_isomeros_3D.sdf 8650
MP7-Q-1005_isomeros_3D.sdf 12775
MP7-Q-1006_isomeros_3D.sdf 9356
MP7-Q-1007_isomeros_3D.sdf 10429
MP7-Q-1008_isomeros_3D.sdf 10405
MP7-Q-1009_isomeros_3D.sdf 12544
MP7-Q-1010_isomeros_3D.sdf 12802
MP7-Q-1011_isomeros_3D.sdf 8350
MP7-Q-1012_isomeros_3D.sdf 10172
MP7-Q-1013_isomeros_3D.sdf 11219
MP7-Q-1014_isomeros_3D.sdf 8666
MP7-Q-1015_isomeros_3D.sdf 8525
MP7-Q-1016_isomeros_3D.sdf 10176
MP7-Q-1017_isomeros_3D.sdf 12554
MP7-Q-1018_isomeros_3D.sdf 9980
MP7-Q-1019_isomeros_3D.sdf 7742
MP7-Q-1020_isomeros_3D.sdf 13111
MP7-Q-1021_isomeros_3D.sdf 27347
MP7-Q-1022_isomeros_3D.sdf 21833
MP7-Q-1023_isomeros_3D.sdf 9031
MP7-Q-1024_isomeros_3D.sdf 15949
MP7-Q-1025_isomeros_3D.sdf 13466
MP7-Q-1026_isomeros_3D.sdf 24392
MP7-Q-1027_isomeros_3D.sdf 17729
MP7-Q-1028_isomeros_3D.sdf 15637
MP7-Q-1029_isomeros_3D.sdf 13095
MP7-Q-1030_isomeros_3D.sdf 11623
MP7-Q-1031_isomeros_3D.sdf 8229
MP7-Q-1032_isomeros_3D.sdf 6920
MP7-Q-1033_isomeros_3D.sdf 7867
MP7-Q-1034_isomeros_3D.sdf 7451
MP7-Q-1035_isomeros_3D.sdf 7609
MP7-Q-1036_isomeros_3D.sdf 7278
MP7-Q-1037_isomeros_3D.sdf 7590
MP7-Q-1038_isomeros_3D.sdf 8187
MP7-Q-1039_isomeros_3D.sdf 16294
MP7-Q-1040_isomeros_3D.sdf 14595
MP7-Q-1041_isomeros_3D.sdf 15775
MP7-Q-1042_isomeros_3D.sdf 14621
MP7-Q-1043_isomeros_3D.sdf 12359
MP7-Q-1044_isomeros_3D.sdf 12644
MP7-Q-1045_isomeros_3D.sdf 8676
MP7-Q-1046_isomeros_3D.sdf 8031
MP7-Q-1047_isomeros_3D.sdf 14346
MP7-Q-1048_isomeros_3D.sdf 11508
MP7-Q-1049_isomeros_3D.sdf 12318
MP7-Q-1050_isomeros_3D.sdf 19609
MP7-Q-1051_isomeros_3D.sdf 7999
MP7-Q-1052_isomeros_3D.sdf 9352
MP7-Q-1053_isomeros_3D.sdf 9193
MP7-Q-1054_isomeros_3D.sdf 9581
MP7-Q-1055_isomeros_3D.sdf 10793
MP7-Q-1056_isomeros_3D.sdf 10252
MP7-Q-1057_isomeros_3D.sdf 9836
MP7-Q-1058_isomeros_3D.sdf 8507
MP7-Q-1059_isomeros_3D.sdf 8118
MP7-Q-1060_isomeros_3D.sdf 9749
MP7-Q-1061_isomeros_3D.sdf 8427
MP7-Q-1062_isomeros_3D.sdf 9802
MP7-Q-1063_isomeros_3D.sdf 9483
MP7-Q-1064_isomeros_3D.sdf 9746
MP7-Q-1065_isomeros_3D.sdf 9303
MP7-Q-1066_isomeros_3D.sdf 8399
MP7-Q-1067_isomeros_3D.sdf 8395
MP7-Q-1068_isomeros_3D.sdf 10914
MP7-Q-1069_isomeros_3D.sdf 9756
MP7-Q-1070_isomeros_3D.sdf 14381
MP7-Q-1071_isomeros_3D.sdf 9726
MP7-Q-1072_isomeros_3D.sdf 5738
MP7-Q-1073_isomeros_3D.sdf 15074
MP7-Q-1074_isomeros_3D.sdf 18623
MP7-Q-1075_isomeros_3D.sdf 19198
MP7-Q-1076_isomeros_3D.sdf 9009
MP7-Q-1077_isomeros_3D.sdf 29015
MP7-Q-1078_isomeros_3D.sdf 21709
MP7-Q-1079_isomeros_3D.sdf 13177
MP7-Q-1080_isomeros_3D.sdf 15926
MP7-Q-1081_isomeros_3D.sdf 13701
MP7-Q-1082_isomeros_3D.sdf 9972
MP7-Q-1083_isomeros_3D.sdf 15818
MP7-Q-1084_isomeros_3D.sdf 8742
MP7-Q-1085_isomeros_3D.sdf 7809
MP7-Q-1086_isomeros_3D.sdf 9219
MP7-Q-1087_isomeros_3D.sdf 7873
MP7-Q-1088_isomeros_3D.sdf 7776
MP7-Q-1089_isomeros_3D.sdf 8945
MP7-Q-1090_isomeros_3D.sdf 8161
MP7-Q-1091_isomeros_3D.sdf 8471
MP7-Q-1092_isomeros_3D.sdf 10584
MP7-Q-1093_isomeros_3D.sdf 12913
MP7-Q-1094_isomeros_3D.sdf 9726
MP7-Q-1095_isomeros_3D.sdf 11352
MP7-Q-1096_isomeros_3D.sdf 8401
MP7-Q-1097_isomeros_3D.sdf 7930
MP7-Q-1098_isomeros_3D.sdf 9775
MP7-Q-1099_isomeros_3D.sdf 7747
MP7-Q-1100_isomeros_3D.sdf 7837
MP7-Q-1101_isomeros_3D.sdf 7782
MP7-Q-1102_isomeros_3D.sdf 8203
MP7-Q-1103_isomeros_3D.sdf 8073
MP7-Q-1104_isomeros_3D.sdf 8019
MP7-Q-1105_isomeros_3D.sdf 12630
MP7-Q-1106_isomeros_3D.sdf 8916
MP7-Q-1107_isomeros_3D.sdf 10044
MP7-Q-1108_isomeros_3D.sdf 7563
MP7-Q-1109_isomeros_3D.sdf 7650
MP7-Q-1110_isomeros_3D.sdf 8136
MP7-Q-1111_isomeros_3D.sdf 11454
MP7-Q-1112_isomeros_3D.sdf 9632
MP7-Q-1113_isomeros_3D.sdf 12476
MP7-Q-1114_isomeros_3D.sdf 10356
MP7-Q-1115_isomeros_3D.sdf 11492
MP7-Q-1116_isomeros_3D.sdf 8974
MP7-Q-1117_isomeros_3D.sdf 11608
MP7-Q-1118_isomeros_3D.sdf 4779
MP7-Q-1119_isomeros_3D.sdf 11809
MP7-Q-1120_isomeros_3D.sdf 9183
MP7-Q-1121_isomeros_3D.sdf 7422
MP7-Q-1122_isomeros_3D.sdf 8486
MP7-Q-1123_isomeros_3D.sdf 12375
MP7-Q-1124_isomeros_3D.sdf 13039
MP7-Q-1125_isomeros_3D.sdf 11202
MP7-Q-1126_isomeros_3D.sdf 10011
MP7-Q-1127_isomeros_3D.sdf 8708
MP7-Q-1128_isomeros_3D.sdf 14116
MP7-Q-1129_isomeros_3D.sdf 13704
MP7-Q-1130_isomeros_3D.sdf 10129
MP7-Q-1131_isomeros_3D.sdf 7015
MP7-Q-1132_isomeros_3D.sdf 8572
MP7-Q-1133_isomeros_3D.sdf 11426
MP7-Q-1134_isomeros_3D.sdf 17151
MP7-Q-1135_isomeros_3D.sdf 13764
MP7-Q-1136_isomeros_3D.sdf 9904
MP7-Q-1137_isomeros_3D.sdf 9965
MP7-Q-1138_isomeros_3D.sdf 10538
MP7-Q-1139_isomeros_3D.sdf 10282
MP7-Q-1140_isomeros_3D.sdf 14710
MP7-Q-1141_isomeros_3D.sdf 7686
MP7-Q-1142_isomeros_3D.sdf 12586
MP7-Q-1143_isomeros_3D.sdf 8296
MP7-Q-1144_isomeros_3D.sdf 10719
MP7-Q-1145_isomeros_3D.sdf 9217
MP7-Q-1146_isomeros_3D.sdf 9106
MP7-Q-1147_isomeros_3D.sdf 8084
MP7-Q-1148_isomeros_3D.sdf 8206
MP7-Q-1149_isomeros_3D.sdf 9808
MP7-Q-1150_isomeros_3D.sdf 14302
MP7-Q-1151_isomeros_3D.sdf 11349
MP7-Q-1152_isomeros_3D.sdf 11752
MP7-Q-1153_isomeros_3D.sdf 10968
MP7-Q-1154_isomeros_3D.sdf 12698
MP7-Q-1155_isomeros_3D.sdf 13810
MP7-Q-1156_isomeros_3D.sdf 13238
MP7-Q-1157_isomeros_3D.sdf 12082
MP7-Q-1158_isomeros_3D.sdf 8305
MP7-Q-1159_isomeros_3D.sdf 11374
MP7-Q-1160_isomeros_3D.sdf 8978
MP7-Q-1161_isomeros_3D.sdf 7736
MP7-Q-1162_isomeros_3D.sdf 11146
MP7-Q-1163_isomeros_3D.sdf 12716
MP7-Q-1164_isomeros_3D.sdf 11556
MP7-Q-1165_isomeros_3D.sdf 14564
MP7-Q-1166_isomeros_3D.sdf 18400
MP7-Q-1167_isomeros_3D.sdf 8477
MP7-Q-1168_isomeros_3D.sdf 8311
MP7-Q-1169_isomeros_3D.sdf 8346
MP7-Q-1170_isomeros_3D.sdf 9132
MP7-Q-1171_isomeros_3D.sdf 8339
MP7-Q-1172_isomeros_3D.sdf 8480
MP7-Q-1173_isomeros_3D.sdf 7568
MP7-Q-1174_isomeros_3D.sdf 10472
MP7-Q-1175_isomeros_3D.sdf 12279
MP7-Q-1176_isomeros_3D.sdf 22754
MP7-Q-1177_isomeros_3D.sdf 8640
MP7-Q-1178_isomeros_3D.sdf 7108
MP7-Q-1179_isomeros_3D.sdf 6319
MP7-Q-1180_isomeros_3D.sdf 6214
MP7-Q-1181_isomeros_3D.sdf 6268
MP7-Q-1182_isomeros_3D.sdf 6294
MP7-Q-1183_isomeros_3D.sdf 6114
MP7-Q-1184_isomeros_3D.sdf 6114
MP7-Q-1185_isomeros_3D.sdf 7275
MP7-Q-1186_isomeros_3D.sdf 8824
MP7-Q-1187_isomeros_3D.sdf 8384
MP7-Q-1188_isomeros_3D.sdf 13810
MP7-Q-1189_isomeros_3D.sdf 13067
MP7-Q-1190_isomeros_3D.sdf 6322
MP7-Q-1191_isomeros_3D.sdf 6252
MP7-Q-1192_isomeros_3D.sdf 6256
MP7-Q-1193_isomeros_3D.sdf 7318
MP7-Q-1194_isomeros_3D.sdf 21535
MP7-Q-1195_isomeros_3D.sdf 10200
MP7-Q-1196_isomeros_3D.sdf 13160
MP7-Q-1197_isomeros_3D.sdf 6702
MP7-Q-1198_isomeros_3D.sdf 6898
MP7-Q-1199_isomeros_3D.sdf 11516
MP7-Q-1200_isomeros_3D.sdf 7147
MP7-Q-1201_isomeros_3D.sdf 7862
MP7-Q-1202_isomeros_3D.sdf 9172
MP7-Q-1203_isomeros_3D.sdf 9263
MP7-Q-1204_isomeros_3D.sdf 9194
MP7-Q-1205_isomeros_3D.sdf 8402
MP7-Q-1206_isomeros_3D.sdf 10842
MP7-Q-1207_isomeros_3D.sdf 9015
MP7-Q-1208_isomeros_3D.sdf 8541
MP7-Q-1209_isomeros_3D.sdf 8164
MP7-Q-1210_isomeros_3D.sdf 8167
MP7-Q-1211_isomeros_3D.sdf 7857
MP7-Q-1212_isomeros_3D.sdf 7978
MP7-Q-1213_isomeros_3D.sdf 9255
MP7-Q-1214_isomeros_3D.sdf 10585
MP7-Q-1215_isomeros_3D.sdf 10058
MP7-Q-1216_isomeros_3D.sdf 9289
MP7-Q-1217_isomeros_3D.sdf 10397
MP7-Q-1218_isomeros_3D.sdf 10027
MP7-Q-1219_isomeros_3D.sdf 6335
MP7-Q-1220_isomeros_3D.sdf 6899
MP7-Q-1221_isomeros_3D.sdf 8137
MP7-Q-1222_isomeros_3D.sdf 9139
MP7-Q-1223_isomeros_3D.sdf 9213
MP7-Q-1224_isomeros_3D.sdf 18604
MP7-Q-1225_isomeros_3D.sdf 7738
MP7-Q-1226_isomeros_3D.sdf 10215
MP7-Q-1227_isomeros_3D.sdf 14438
MP7-Q-1228_isomeros_3D.sdf 11723
MP7-Q-1229_isomeros_3D.sdf 16619
MP7-Q-1230_isomeros_3D.sdf 9308
MP7-Q-1231_isomeros_3D.sdf 7731
MP7-Q-1232_isomeros_3D.sdf 7295
MP7-Q-1233_isomeros_3D.sdf 7294
MP7-Q-1234_isomeros_3D.sdf 7219
MP7-Q-1235_isomeros_3D.sdf 14903
MP7-Q-1236_isomeros_3D.sdf 17192
MP7-Q-1237_isomeros_3D.sdf 17533
MP7-Q-1238_isomeros_3D.sdf 9231
MP7-Q-1239_isomeros_3D.sdf 15948
MP7-Q-1240_isomeros_3D.sdf 12820
MP7-Q-1241_isomeros_3D.sdf 11368
MP7-Q-1242_isomeros_3D.sdf 16197
MP7-Q-1243_isomeros_3D.sdf 13029
MP7-Q-1244_isomeros_3D.sdf 8908
MP7-Q-1245_isomeros_3D.sdf 11545
MP7-Q-1246_isomeros_3D.sdf 9614
MP7-Q-1247_isomeros_3D.sdf 10120
MP7-Q-1248_isomeros_3D.sdf 11644
MP7-Q-1249_isomeros_3D.sdf 10257
MP7-Q-1250_isomeros_3D.sdf 8030
MP7-Q-1251_isomeros_3D.sdf 12267
MP7-Q-1252_isomeros_3D.sdf 21887
MP7-Q-1253_isomeros_3D.sdf 10105
MP7-Q-1254_isomeros_3D.sdf 7964
MP7-Q-1255_isomeros_3D.sdf 10193
MP7-Q-1256_isomeros_3D.sdf 10124
MP7-Q-1257_isomeros_3D.sdf 9304
MP7-Q-1258_isomeros_3D.sdf 10069
MP7-Q-1259_isomeros_3D.sdf 10361
MP7-Q-1260_isomeros_3D.sdf 10372
MP7-Q-1261_isomeros_3D.sdf 9730
MP7-Q-1262_isomeros_3D.sdf 11210
MP7-Q-1263_isomeros_3D.sdf 9712
MP7-Q-1264_isomeros_3D.sdf 10083
MP7-Q-1265_isomeros_3D.sdf 10862
MP7-Q-1266_isomeros_3D.sdf 10986
MP7-Q-1267_isomeros_3D.sdf 11604
MP7-Q-1268_isomeros_3D.sdf 12415
MP7-Q-1269_isomeros_3D.sdf 12683
MP7-Q-1270_isomeros_3D.sdf 13307
MP7-Q-1271_isomeros_3D.sdf 13145
MP7-Q-1272_isomeros_3D.sdf 8485
MP7-Q-1273_isomeros_3D.sdf 9300
MP7-Q-1274_isomeros_3D.sdf 7386
MP7-Q-1275_isomeros_3D.sdf 7790
MP7-Q-1276_isomeros_3D.sdf 7652
MP7-Q-1277_isomeros_3D.sdf 7886
MP7-Q-1278_isomeros_3D.sdf 7559
MP7-Q-1279_isomeros_3D.sdf 11212
MP7-Q-1280_isomeros_3D.sdf 7453
MP7-Q-1281_isomeros_3D.sdf 11126
MP7-Q-1282_isomeros_3D.sdf 7325
MP7-Q-1283_isomeros_3D.sdf 10132
MP7-Q-1284_isomeros_3D.sdf 13841
MP7-Q-1285_isomeros_3D.sdf 8855
MP7-Q-1286_isomeros_3D.sdf 12048
MP7-Q-1287_isomeros_3D.sdf 9596
MP7-Q-1288_isomeros_3D.sdf 7367
MP7-Q-1289_isomeros_3D.sdf 6533
MP7-Q-1290_isomeros_3D.sdf 7799
MP7-Q-1291_isomeros_3D.sdf 10269
MP7-Q-1292_isomeros_3D.sdf 9484
MP7-Q-1293_isomeros_3D.sdf 9924
MP7-Q-1294_isomeros_3D.sdf 7915
MP7-Q-1295_isomeros_3D.sdf 7206
MP7-Q-1296_isomeros_3D.sdf 9016
MP7-Q-1297_isomeros_3D.sdf 8086
MP7-Q-1298_isomeros_3D.sdf 14262
MP7-Q-1299_isomeros_3D.sdf 9669
MP7-Q-1300_isomeros_3D.sdf 9826
MP7-Q-1301_isomeros_3D.sdf 10625
MP7-Q-1302_isomeros_3D.sdf 9636
MP7-Q-1303_isomeros_3D.sdf 10565
MP7-Q-1304_isomeros_3D.sdf 9977
MP7-Q-1305_isomeros_3D.sdf 10068
MP7-Q-1306_isomeros_3D.sdf 12302
MP7-Q-1307_isomeros_3D.sdf 12194
MP7-Q-1308_isomeros_3D.sdf 11687
MP7-Q-1309_isomeros_3D.sdf 9430
MP7-Q-1310_isomeros_3D.sdf 9587
MP7-Q-1311_isomeros_3D.sdf 11094
MP7-Q-1312_isomeros_3D.sdf 11887
MP7-Q-1313_isomeros_3D.sdf 9245
MP7-Q-1314_isomeros_3D.sdf 8399
MP7-Q-1315_isomeros_3D.sdf 9822
MP7-Q-1316_isomeros_3D.sdf 8217
MP7-Q-1317_isomeros_3D.sdf 9764
MP7-Q-1318_isomeros_3D.sdf 11611
MP7-Q-1319_isomeros_3D.sdf 9308
MP7-Q-1320_isomeros_3D.sdf 9422
MP7-Q-1321_isomeros_3D.sdf 10434
MP7-Q-1322_isomeros_3D.sdf 10544
MP7-Q-1323_isomeros_3D.sdf 11450
MP7-Q-1324_isomeros_3D.sdf 11016
MP7-Q-1325_isomeros_3D.sdf 9710
MP7-Q-1326_isomeros_3D.sdf 9516
MP7-Q-1327_isomeros_3D.sdf 9739
MP7-Q-1328_isomeros_3D.sdf 8037
MP7-Q-1329_isomeros_3D.sdf 10240
MP7-Q-1330_isomeros_3D.sdf 9117
MP7-Q-1331_isomeros_3D.sdf 9789
MP7-Q-1332_isomeros_3D.sdf 9428
MP7-Q-1333_isomeros_3D.sdf 9817
MP7-Q-1334_isomeros_3D.sdf 10677
MP7-Q-1335_isomeros_3D.sdf 10327
MP7-Q-1336_isomeros_3D.sdf 10971
MP7-Q-1337_isomeros_3D.sdf 13374
MP7-Q-1338_isomeros_3D.sdf 9157
MP7-Q-1339_isomeros_3D.sdf 11776
MP7-Q-1340_isomeros_3D.sdf 9843
MP7-Q-1341_isomeros_3D.sdf 10676
MP7-Q-1342_isomeros_3D.sdf 8589
MP7-Q-1343_isomeros_3D.sdf 9690
MP7-Q-1344_isomeros_3D.sdf 10752
MP7-Q-1345_isomeros_3D.sdf 9074
MP7-Q-1346_isomeros_3D.sdf 9311
MP7-Q-1347_isomeros_3D.sdf 11138
MP7-Q-1348_isomeros_3D.sdf 7868
MP7-Q-1349_isomeros_3D.sdf 8971
MP7-Q-1350_isomeros_3D.sdf 6713
MP7-Q-1351_isomeros_3D.sdf 6650
MP7-Q-1352_isomeros_3D.sdf 8952
MP7-Q-1353_isomeros_3D.sdf 9719
MP7-Q-1354_isomeros_3D.sdf 10127
MP7-Q-1355_isomeros_3D.sdf 12167
MP7-Q-1356_isomeros_3D.sdf 8679
MP7-Q-1357_isomeros_3D.sdf 7804
MP7-Q-1358_isomeros_3D.sdf 8314
MP7-Q-1359_isomeros_3D.sdf 7835
MP7-Q-1360_isomeros_3D.sdf 4361
MP7-Q-1361_isomeros_3D.sdf 8692
MP7-Q-1362_isomeros_3D.sdf 9095
MP7-Q-1363_isomeros_3D.sdf 10226
MP7-Q-1364_isomeros_3D.sdf 9531
MP7-Q-1365_isomeros_3D.sdf 10044
MP7-Q-1366_isomeros_3D.sdf 8425
MP7-Q-1367_isomeros_3D.sdf 8085
MP7-Q-1368_isomeros_3D.sdf 9022
MP7-Q-1369_isomeros_3D.sdf 10815
MP7-Q-1370_isomeros_3D.sdf 20962
MP7-Q-1371_isomeros_3D.sdf 6718
MP7-Q-1372_isomeros_3D.sdf 17080
MP7-Q-1373_isomeros_3D.sdf 8322
MP7-Q-1374_isomeros_3D.sdf 3685
MP7-Q-1375_isomeros_3D.sdf 10078
MP7-Q-1376_isomeros_3D.sdf 8005
MP7-Q-1377_isomeros_3D.sdf 7978
MP7-Q-1378_isomeros_3D.sdf 7825
MP7-Q-1379_isomeros_3D.sdf 8910
MP7-Q-1380_isomeros_3D.sdf 11096
MP7-Q-1381_isomeros_3D.sdf 9214
MP7-Q-1382_isomeros_3D.sdf 8915
MP7-Q-1383_isomeros_3D.sdf 9375
MP7-Q-1384_isomeros_3D.sdf 11348
MP7-Q-1385_isomeros_3D.sdf 11413
MP7-Q-1386_isomeros_3D.sdf 10907
MP7-Q-1387_isomeros_3D.sdf 9322
MP7-Q-1388_isomeros_3D.sdf 9619
MP7-Q-1389_isomeros_3D.sdf 12083
MP7-Q-1390_isomeros_3D.sdf 9531
MP7-Q-1391_isomeros_3D.sdf 11439
MP7-Q-1392_isomeros_3D.sdf 11518
MP7-Q-1393_isomeros_3D.sdf 10445
MP7-Q-1394_isomeros_3D.sdf 42960
MP7-Q-1395_isomeros_3D.sdf 8498
MP7-Q-1396_isomeros_3D.sdf 9732
MP7-Q-1397_isomeros_3D.sdf 11276
MP7-Q-1398_isomeros_3D.sdf 12405
MP7-Q-1399_isomeros_3D.sdf 8039
MP7-Q-1400_isomeros_3D.sdf 8544
MP7-Q-1401_isomeros_3D.sdf 46329
MP7-Q-1402_isomeros_3D.sdf 10099
MP7-Q-1403_isomeros_3D.sdf 6367
MP7-Q-1404_isomeros_3D.sdf 7070
MP7-Q-1405_isomeros_3D.sdf 9862
MP7-Q-1406_isomeros_3D.sdf 10046
MP7-Q-1407_isomeros_3D.sdf 8722
MP7-Q-1408_isomeros_3D.sdf 7562
MP7-Q-1409_isomeros_3D.sdf 20264
MP7-Q-1410_isomeros_3D.sdf 8083
MP7-Q-1411_isomeros_3D.sdf 9898
MP7-Q-1412_isomeros_3D.sdf 11338
MP7-Q-1413_isomeros_3D.sdf 1370
MP7-Q-1414_isomeros_3D.sdf 9559
MP7-Q-1415_isomeros_3D.sdf 10394
MP7-Q-1416_isomeros_3D.sdf 10932
MP7-Q-1417_isomeros_3D.sdf 12402
MP7-Q-1418_isomeros_3D.sdf 14443
MP7-Q-1419_isomeros_3D.sdf 1362
MP7-Q-1420_isomeros_3D.sdf 6333
MP7-Q-1421_isomeros_3D.sdf 6018
MP7-Q-1422_isomeros_3D.sdf 7218
MP7-Q-1423_isomeros_3D.sdf 7532
MP7-Q-1424_isomeros_3D.sdf 13440
MP7-Q-1425_isomeros_3D.sdf 9988
MP7-Q-1426_isomeros_3D.sdf 20054
MP7-Q-1427_isomeros_3D.sdf 25788
MP7-Q-1428_isomeros_3D.sdf 29909
MP7-Q-1429_isomeros_3D.sdf 18938
MP7-Q-1430_isomeros_3D.sdf 11538
MP7-Q-1431_isomeros_3D.sdf 11752
MP7-Q-1432_isomeros_3D.sdf 9551
MP7-Q-1433_isomeros_3D.sdf 21276
MP7-Q-1434_isomeros_3D.sdf 25924
MP7-Q-1435_isomeros_3D.sdf 13179
MP7-Q-1436_isomeros_3D.sdf 15400
MP7-Q-1437_isomeros_3D.sdf 10287
MP7-Q-1438_isomeros_3D.sdf 27673
MP7-Q-1439_isomeros_3D.sdf 22360
MP7-Q-1440_isomeros_3D.sdf 13920
MP7-Q-1441_isomeros_3D.sdf 12196
MP7-Q-1442_isomeros_3D.sdf 15204
MP7-Q-1443_isomeros_3D.sdf 15593
MP7-Q-1444_isomeros_3D.sdf 11632
MP7-Q-1445_isomeros_3D.sdf 12510
MP7-Q-1446_isomeros_3D.sdf 13411
MP7-Q-1447_isomeros_3D.sdf 12772
MP7-Q-1448_isomeros_3D.sdf 11497
MP7-Q-1449_isomeros_3D.sdf 13946
MP7-Q-1450_isomeros_3D.sdf 10922
MP7-Q-1451_isomeros_3D.sdf 12508
MP7-Q-1452_isomeros_3D.sdf 13904
MP7-Q-1453_isomeros_3D.sdf 12858
MP7-Q-1454_isomeros_3D.sdf 11390
MP7-Q-1455_isomeros_3D.sdf 12298
MP7-Q-1456_isomeros_3D.sdf 13056
MP7-Q-1457_isomeros_3D.sdf 12589
MP7-Q-1458_isomeros_3D.sdf 13745
MP7-Q-1459_isomeros_3D.sdf 13356
MP7-Q-1460_isomeros_3D.sdf 11053
MP7-Q-1461_isomeros_3D.sdf 12444
MP7-Q-1462_isomeros_3D.sdf 13142
MP7-Q-1463_isomeros_3D.sdf 9498
MP7-Q-1464_isomeros_3D.sdf 10347
MP7-Q-1465_isomeros_3D.sdf 9417
MP7-Q-1466_isomeros_3D.sdf 7987
MP7-Q-1467_isomeros_3D.sdf 8341
MP7-Q-1468_isomeros_3D.sdf 8983
MP7-Q-1469_isomeros_3D.sdf 13627
MP7-Q-1470_isomeros_3D.sdf 10989
MP7-Q-1471_isomeros_3D.sdf 13559
MP7-Q-1472_isomeros_3D.sdf 13414
MP7-Q-1473_isomeros_3D.sdf 20068
MP7-Q-1474_isomeros_3D.sdf 9599
MP7-Q-1475_isomeros_3D.sdf 10903
MP7-Q-1476_isomeros_3D.sdf 21653
MP7-Q-1477_isomeros_3D.sdf 8077
MP7-Q-1478_isomeros_3D.sdf 10088
MP7-Q-1479_isomeros_3D.sdf 8046
MP7-Q-1480_isomeros_3D.sdf 10332
MP7-Q-1481_isomeros_3D.sdf 11895
MP7-Q-1482_isomeros_3D.sdf 12267
MP7-Q-1483_isomeros_3D.sdf 821
MP7-Q-1484_isomeros_3D.sdf 10866
MP7-Q-1485_isomeros_3D.sdf 32779
MP7-Q-1486_isomeros_3D.sdf 796
MP7-Q-1487_isomeros_3D.sdf 18674
MP7-Q-1488_isomeros_3D.sdf 16735
MP7-Q-1489_isomeros_3D.sdf 10183
MP7-Q-1490_isomeros_3D.sdf 17031
MP7-Q-1491_isomeros_3D.sdf 10359
MP7-Q-1492_isomeros_3D.sdf 10180
MP7-Q-1493_isomeros_3D.sdf 10059
MP7-Q-1494_isomeros_3D.sdf 11880
MP7-Q-1495_isomeros_3D.sdf 10890
MP7-Q-1496_isomeros_3D.sdf 11081
MP7-Q-1497_isomeros_3D.sdf 15272
MP7-Q-1498_isomeros_3D.sdf 12560
MP7-Q-1499_isomeros_3D.sdf 10972
MP7-Q-1500_isomeros_3D.sdf 11191
MP7-Q-1501_isomeros_3D.sdf 18482
MP7-Q-1502_isomeros_3D.sdf 18332
MP7-Q-1503_isomeros_3D.sdf 18260
MP7-Q-1504_isomeros_3D.sdf 18235
MP7-Q-1505_isomeros_3D.sdf 13007
MP7-Q-1506_isomeros_3D.sdf 9927
MP7-Q-1507_isomeros_3D.sdf 27204
MP7-Q-1508_isomeros_3D.sdf 15979
MP7-Q-1509_isomeros_3D.sdf 15497
MP7-Q-1510_isomeros_3D.sdf 8500
MP7-Q-1511_isomeros_3D.sdf 10492
MP7-Q-1512_isomeros_3D.sdf 11192
MP7-Q-1513_isomeros_3D.sdf 12072
MP7-Q-1514_isomeros_3D.sdf 16418
MP7-Q-1515_isomeros_3D.sdf 17680
MP7-Q-1516_isomeros_3D.sdf 11532
MP7-Q-1517_isomeros_3D.sdf 15488
MP7-Q-1518_isomeros_3D.sdf 21966
MP7-Q-1519_isomeros_3D.sdf 15117
MP7-Q-1520_isomeros_3D.sdf 6692
MP7-Q-1521_isomeros_3D.sdf 7540
MP7-Q-1522_isomeros_3D.sdf 8087
MP7-Q-1523_isomeros_3D.sdf 7220
MP7-Q-1524_isomeros_3D.sdf 7048
MP7-Q-1525_isomeros_3D.sdf 6994
MP7-Q-1526_isomeros_3D.sdf 7893
MP7-Q-1527_isomeros_3D.sdf 10334
MP7-Q-1528_isomeros_3D.sdf 9731
MP7-Q-1529_isomeros_3D.sdf 8415
MP7-Q-1530_isomeros_3D.sdf 9749
MP7-Q-1531_isomeros_3D.sdf 6554
MP7-Q-1532_isomeros_3D.sdf 7312
MP7-Q-1533_isomeros_3D.sdf 6218
MP7-Q-1534_isomeros_3D.sdf 6248
MP7-Q-1535_isomeros_3D.sdf 6524
MP7-Q-1536_isomeros_3D.sdf 6942
MP7-Q-1537_isomeros_3D.sdf 7360
MP7-Q-1538_isomeros_3D.sdf 6994
MP7-Q-1539_isomeros_3D.sdf 6963
MP7-Q-1540_isomeros_3D.sdf 7550
MP7-Q-1541_isomeros_3D.sdf 6392
MP7-Q-1542_isomeros_3D.sdf 6116
MP7-Q-1543_isomeros_3D.sdf 6520
MP7-Q-1544_isomeros_3D.sdf 6114
MP7-Q-1545_isomeros_3D.sdf 6112
MP7-Q-1546_isomeros_3D.sdf 6628
MP7-Q-1547_isomeros_3D.sdf 6500
MP7-Q-1548_isomeros_3D.sdf 6468
MP7-Q-1549_isomeros_3D.sdf 6884
MP7-Q-1550_isomeros_3D.sdf 6458
MP7-Q-1551_isomeros_3D.sdf 6346
MP7-Q-1552_isomeros_3D.sdf 28725
MP7-Q-1553_isomeros_3D.sdf 8369
MP7-Q-1554_isomeros_3D.sdf 8127
MP7-Q-1555_isomeros_3D.sdf 8048
MP7-Q-1556_isomeros_3D.sdf 8207
MP7-Q-1557_isomeros_3D.sdf 8088
MP7-Q-1558_isomeros_3D.sdf 7816
MP7-Q-1559_isomeros_3D.sdf 8092
MP7-Q-1560_isomeros_3D.sdf 7942
MP7-Q-1561_isomeros_3D.sdf 7604
MP7-Q-1562_isomeros_3D.sdf 7638
MP7-Q-1563_isomeros_3D.sdf 7602
MP7-Q-1564_isomeros_3D.sdf 7486
MP7-Q-1565_isomeros_3D.sdf 7392
MP7-Q-1566_isomeros_3D.sdf 7452
MP7-Q-1567_isomeros_3D.sdf 7123
MP7-Q-1568_isomeros_3D.sdf 7358
MP7-Q-1569_isomeros_3D.sdf 7101
MP7-Q-1570_isomeros_3D.sdf 7120
MP7-Q-1571_isomeros_3D.sdf 7365
MP7-Q-1572_isomeros_3D.sdf 7210
MP7-Q-1573_isomeros_3D.sdf 7181
MP7-Q-1574_isomeros_3D.sdf 7178
MP7-Q-1575_isomeros_3D.sdf 7262
MP7-Q-1576_isomeros_3D.sdf 7303
MP7-Q-1577_isomeros_3D.sdf 7389
MP7-Q-1578_isomeros_3D.sdf 8595
MP7-Q-1579_isomeros_3D.sdf 7398
MP7-Q-1580_isomeros_3D.sdf 7359
MP7-Q-1581_isomeros_3D.sdf 7308
MP7-Q-1582_isomeros_3D.sdf 7335
MP7-Q-1583_isomeros_3D.sdf 7179
MP7-Q-1584_isomeros_3D.sdf 7206
MP7-Q-1585_isomeros_3D.sdf 7288
MP7-Q-1586_isomeros_3D.sdf 7217
MP7-Q-1587_isomeros_3D.sdf 7258
MP7-Q-1588_isomeros_3D.sdf 7268
MP7-Q-1589_isomeros_3D.sdf 7229
MP7-Q-1590_isomeros_3D.sdf 7112
MP7-Q-1591_isomeros_3D.sdf 6939
MP7-Q-1592_isomeros_3D.sdf 7085
MP7-Q-1593_isomeros_3D.sdf 6926
MP7-Q-1594_isomeros_3D.sdf 7015
MP7-Q-1595_isomeros_3D.sdf 6964
MP7-Q-1596_isomeros_3D.sdf 7026
MP7-Q-1597_isomeros_3D.sdf 6920
MP7-Q-1598_isomeros_3D.sdf 6866
MP7-Q-1599_isomeros_3D.sdf 6995
MP7-Q-1600_isomeros_3D.sdf 7068
MP7-Q-1601_isomeros_3D.sdf 6877
MP7-Q-1602_isomeros_3D.sdf 6876
MP7-Q-1603_isomeros_3D.sdf 6806
MP7-Q-1604_isomeros_3D.sdf 6955
MP7-Q-1605_isomeros_3D.sdf 6899
MP7-Q-1606_isomeros_3D.sdf 6908
MP7-Q-1607_isomeros_3D.sdf 6888
MP7-Q-1608_isomeros_3D.sdf 7001
MP7-Q-1609_isomeros_3D.sdf 6921
MP7-Q-1610_isomeros_3D.sdf 6989
MP7-Q-1611_isomeros_3D.sdf 7013
MP7-Q-1612_isomeros_3D.sdf 7093
MP7-Q-1613_isomeros_3D.sdf 6999
MP7-Q-1614_isomeros_3D.sdf 6913
MP7-Q-1615_isomeros_3D.sdf 7027
MP7-Q-1616_isomeros_3D.sdf 7113
MP7-Q-1617_isomeros_3D.sdf 7024
MP7-Q-1618_isomeros_3D.sdf 6929
MP7-Q-1619_isomeros_3D.sdf 6909
MP7-Q-1620_isomeros_3D.sdf 6895
MP7-Q-1621_isomeros_3D.sdf 6827
MP7-Q-1622_isomeros_3D.sdf 6747
MP7-Q-1623_isomeros_3D.sdf 6697
MP7-Q-1624_isomeros_3D.sdf 6767
MP7-Q-1625_isomeros_3D.sdf 6806
MP7-Q-1626_isomeros_3D.sdf 6747
MP7-Q-1627_isomeros_3D.sdf 6799
MP7-Q-1628_isomeros_3D.sdf 6939
MP7-Q-1629_isomeros_3D.sdf 6837
MP7-Q-1630_isomeros_3D.sdf 6931
MP7-Q-1631_isomeros_3D.sdf 6932
MP7-Q-1632_isomeros_3D.sdf 6852
MP7-Q-1633_isomeros_3D.sdf 6835
MP7-Q-1634_isomeros_3D.sdf 6853
MP7-Q-1635_isomeros_3D.sdf 6885
MP7-Q-1636_isomeros_3D.sdf 6728
MP7-Q-1637_isomeros_3D.sdf 6676
MP7-Q-1638_isomeros_3D.sdf 6863
MP7-Q-1639_isomeros_3D.sdf 7359
MP7-Q-1640_isomeros_3D.sdf 7677
MP7-Q-1641_isomeros_3D.sdf 7560
MP7-Q-1642_isomeros_3D.sdf 7607
MP7-Q-1643_isomeros_3D.sdf 7492
MP7-Q-1644_isomeros_3D.sdf 7413
MP7-Q-1645_isomeros_3D.sdf 7462
MP7-Q-1646_isomeros_3D.sdf 7700
MP7-Q-1647_isomeros_3D.sdf 7317
MP7-Q-1648_isomeros_3D.sdf 7362
MP7-Q-1649_isomeros_3D.sdf 7029
MP7-Q-1650_isomeros_3D.sdf 6938
MP7-Q-1651_isomeros_3D.sdf 8544
MP7-Q-1652_isomeros_3D.sdf 13385
MP7-Q-1653_isomeros_3D.sdf 15499
MP7-Q-1654_isomeros_3D.sdf 14036
MP7-Q-1655_isomeros_3D.sdf 14378
MP7-Q-1656_isomeros_3D.sdf 15352
MP7-Q-1657_isomeros_3D.sdf 5828
MP7-Q-1658_isomeros_3D.sdf 12661
MP7-Q-1659_isomeros_3D.sdf 11903
MP7-Q-1660_isomeros_3D.sdf 11462
MP7-Q-1661_isomeros_3D.sdf 2811
MP7-Q-1662_isomeros_3D.sdf 12193
MP7-Q-1663_isomeros_3D.sdf 17394
MP7-Q-1664_isomeros_3D.sdf 5476
MP7-Q-1665_isomeros_3D.sdf 18758
MP7-Q-1666_isomeros_3D.sdf 20820
MP7-Q-1667_isomeros_3D.sdf 16526
MP7-Q-1668_isomeros_3D.sdf 16804
MP7-Q-1669_isomeros_3D.sdf 15766
MP7-Q-1670_isomeros_3D.sdf 14848
MP7-Q-1671_isomeros_3D.sdf 7528
MP7-Q-1672_isomeros_3D.sdf 13671
MP7-Q-1673_isomeros_3D.sdf 14083
MP7-Q-1674_isomeros_3D.sdf 21028
MP7-Q-1675_isomeros_3D.sdf 20889
MP7-Q-1676_isomeros_3D.sdf 18773
MP7-Q-1677_isomeros_3D.sdf 13849
MP7-Q-1678_isomeros_3D.sdf 16186
MP7-Q-1679_isomeros_3D.sdf 22234
TOTAL 17,641,929