Creación de bibliotecas combinatorias partiendo de fragmentos generados por 'scaffold-hopping'.
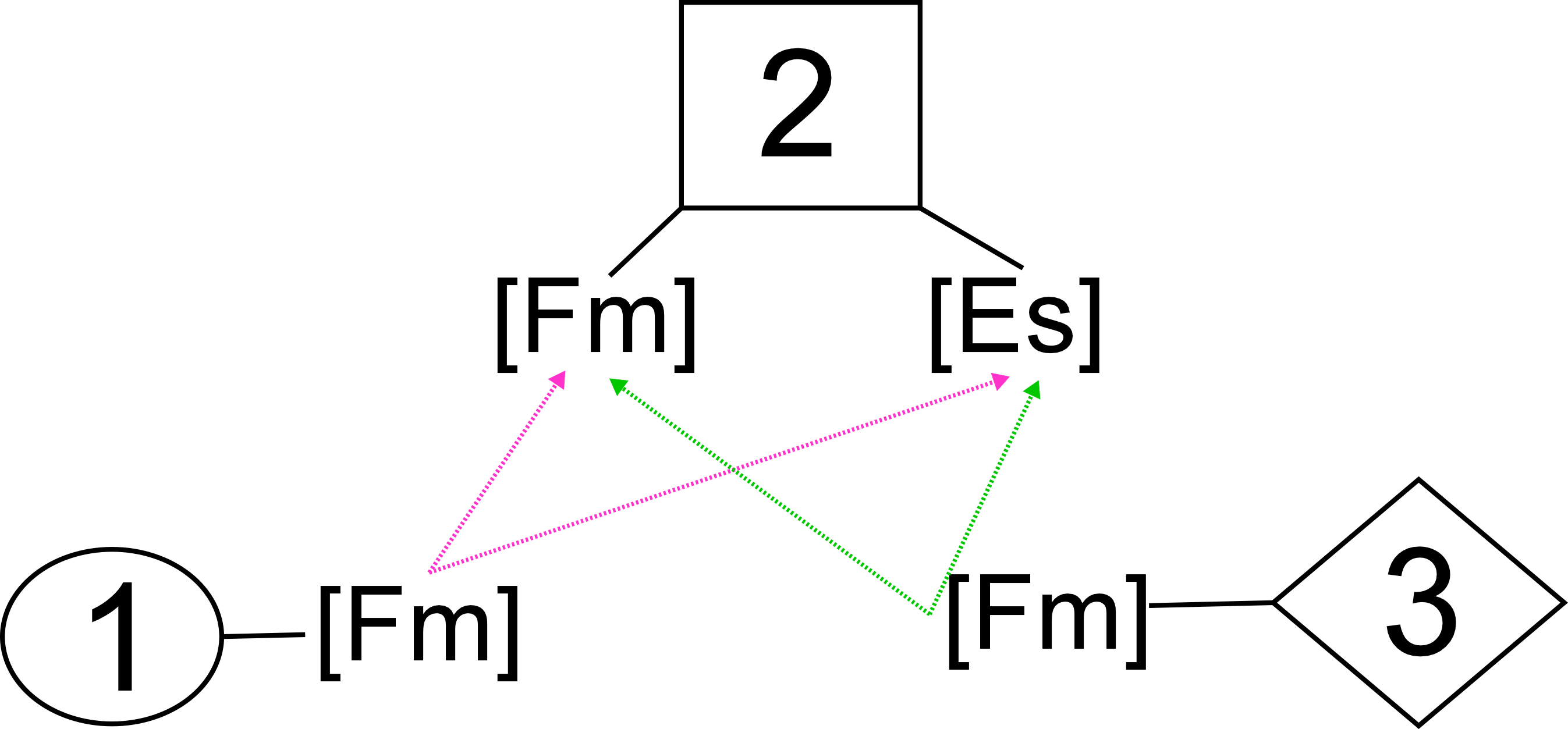
El siguiente script de Python usa RDkit y crea todas las combinaciones de 3 tipos de fragmentos de moléculas orgánicas usando un fichero C:\Users\jant\Desktop\notebook\combina-245.txt que tiene separadas por tabulabores tres columnas con los SMILES de los tres fragmentos a combinar. Los puntos de unión por enlace covalente de cada fragmento están escritos en cada SMILES como [Fm], de forma que Fragment-UNO tiene un solo sitio de union con Fragment-DOS. Fragment-DOS tiene dos sitios de conexión identificados como [Fm] y [Es]. Fragment-UNO puede conectarse a Fragment-DOS a través de su sitio [Fm] o por el sitio [Es]. Finalmente Fragment-TRES tiene solo un sitio de conexión llamado [Fm] y solo puede conectarse con Fragment-DOS, tanto por el sitio de unión covalente [Fm] o por el sitio [Es].
La razón de ser de estas combinaciones tiene que ver con el punto de partida, que no es otro que la evolución de un compuesto 'hit' (con actividad biológica alta pero una solubilidad en agua que queremos mejorar, junto con propiedades ADMET más favorables para DILI, Ames y hERG).
Asegúrate de que el archivo C:\Users\jant\Desktop\notebook\combina-245.txt no tenga encabezados.
Como el Fragment-DOS tiene dos sitios, el script generará:
Fragment-UNOen [Fm] deFragment-DOSyFragment-TRESen [Es] deFragment-DOS.Fragment-UNOen [Es] deFragment-DOSyFragment-TRESen [Fm] deFragment-DOS.
Cuando se trabaja con librerías combinatorias, la simetría es el factor que más "infla" los archivos innecesariamente. Si el Fragmento 2 es simétrico (por ejemplo, si los puntos [Fm] y [Es] están en posiciones equivalentes de un anillo de benceno), las opciones A y B generarán la misma molécula.
Necesitamos un segundo script de filtrado para que sea extremadamente eficiente en memoria. En lugar de cargar los 1.7 millones de SMILES en una lista, utilizaremos un set de InChIKeys. El InChIKey es una huella digital (hash) de 27 caracteres fija. Es mucho más corto que un SMILES largo y permite comparar si dos moléculas son idénticas estructuralmente (incluyendo estereoquímica si la hubiera) de forma casi instantánea.
Primer script...
from rdkit import Chem
from rdkit.Chem import AllChem
import pandas as pd
import itertools
import sys
# --- Configuración de Reacciones ---
# Definimos los SMARTS para unir por [Fm] (Fermio) y [Es] (Einstenio)
rxn_fm = AllChem.ReactionFromSmarts('[*:1][Fm].[Fm][*:2]>>[*:1][*:2]')
rxn_es = AllChem.ReactionFromSmarts('[*:1][Es].[Es][*:2]>>[*:1][*:2]')
def combine_fragments(f1_mol, f2_mol, f3_mol, f1_smi_raw, f3_smi_raw):
"""
Realiza las dos combinaciones posibles:
1. F1 en sitio [Fm] de F2 y F3 en sitio [Es] de F2
2. F1 en sitio [Es] de F2 y F3 en sitio [Fm] de F2
"""
results = set()
# OPCIÓN A: F1-[Fm] + F2-[Fm] -> Intermedio-[Es] + F3-[Es]
# Cambiamos etiqueta de F3 para que encaje en el sitio Es de F2
f3_es = Chem.MolFromSmiles(f3_smi_raw.replace("[Fm]", "[Es]"))
if f3_es:
p1 = rxn_fm.RunReactants((f1_mol, f2_mol))
if p1:
p2 = rxn_es.RunReactants((p1[0][0], f3_es))
if p2:
results.add(Chem.MolToSmiles(p2[0][0]))
# OPCIÓN B: F1-[Es] + F2-[Es] -> Intermedio-[Fm] + F3-[Fm]
# Cambiamos etiqueta de F1 para que encaje en el sitio Es de F2
f1_es = Chem.MolFromSmiles(f1_smi_raw.replace("[Fm]", "[Es]"))
if f1_es:
p1 = rxn_es.RunReactants((f1_es, f2_mol))
if p1:
p2 = rxn_fm.RunReactants((p1[0][0], f3_mol))
if p2:
results.add(Chem.MolToSmiles(p2[0][0]))
return results
# --- Rutas de archivos ---
input_path = r'C:\Users\jant\Desktop\notebook\combina-245.txt'
output_path = r'C:\Users\jant\Desktop\notebook\biblioteca_completa.txt'
# --- Carga y Limpieza ---
try:
print("Leyendo archivo de fragmentos...")
df_raw = pd.read_csv(input_path, sep='\t', header=None, dtype=str)
# Extraer columnas ignorando celdas vacías (para manejar longitudes distintas)
list_f1 = df_raw[0].dropna().unique().tolist()
list_f2 = df_raw[1].dropna().unique().tolist()
list_f3 = df_raw[2].dropna().unique().tolist()
print(f"Fragmentos cargados: F1={len(list_f1)}, F2={len(list_f2)}, F3={len(list_f3)}")
# Pre-conversión a objetos Mol para optimizar el bucle
mols_f1 = [(Chem.MolFromSmiles(s), s) for s in list_f1 if Chem.MolFromSmiles(s)]
mols_f2 = [Chem.MolFromSmiles(s) for s in list_f2 if Chem.MolFromSmiles(s)]
mols_f3 = [(Chem.MolFromSmiles(s), s) for s in list_f3 if Chem.MolFromSmiles(s)]
total_iteraciones = len(mols_f1) * len(mols_f2) * len(mols_f3)
print(f"Total de tríos a procesar: {total_iteraciones}")
print("-" * 30)
# --- Generación con seguimiento de progreso ---
count_prod = 0
count_iter = 0
diez_por_ciento = total_iteraciones // 10 if total_iteraciones > 10 else 1
with open(output_path, 'w') as f:
f.write("SMILES\n")
# itertools.product crea el producto cartesiano eficientemente
for (f1_m, f1_s), f2_m, (f3_m, f3_s) in itertools.product(mols_f1, mols_f2, mols_f3):
productos = combine_fragments(f1_m, f2_m, f3_m, f1_s, f3_s)
for p in productos:
f.write(f"{p}\n")
count_prod += 1
count_iter += 1
# Notificación de progreso
if count_iter % diez_por_ciento == 0:
progreso = (count_iter / total_iteraciones) * 100
print(f"Progreso: {progreso:.0f}% completado ({count_iter} tríos procesados)...")
sys.stdout.flush() # Forzar actualización en consola
print("-" * 30)
print(f"¡Finalizado con éxito!")
print(f"Moléculas totales generadas y guardadas: {count_prod}")
print(f"Archivo resultante: {output_path}")
except Exception as e:
print(f"Error durante la ejecución: {e}")
Segundo script...
from rdkit import Chem
import os
import sys
# --- Configuración de rutas ---
input_path = r'C:\Users\jant\Desktop\notebook\biblioteca_completa.txt'
output_clean_path = r'C:\Users\jant\Desktop\notebook\biblioteca_UNICA_InChIKeys.txt'
def filtrar_duplicados_con_progreso(input_file, output_file):
if not os.path.exists(input_file):
print(f"Error: No se encuentra el archivo en {input_file}")
return
# 1. Contar líneas totales para calcular el progreso (rápido)
print("Calculando tamaño del archivo para la barra de progreso...")
with open(input_file, 'r') as f:
total_lineas = sum(1 for line in f) - 1 # Restamos la cabecera
# Intervalo para avisar cada 10%
diez_por_ciento = total_lineas // 10 if total_lineas > 10 else 1
vistas = set()
count_total = 0
count_unicas = 0
print(f"Iniciando limpieza de {total_lineas} moléculas...")
print("-" * 30)
with open(input_file, 'r') as f_in, open(output_file, 'w') as f_out:
f_in.readline() # Saltamos la cabecera "SMILES"
f_out.write("SMILES,InChIKey\n")
for line in f_in:
smi = line.strip()
if not smi:
continue
count_total += 1
# Convertimos a Mol para estandarizar y luego a InChIKey
mol = Chem.MolFromSmiles(smi)
if mol:
# El InChIKey es la forma más segura de detectar si dos estructuras son la misma
ikey = Chem.MolToInchiKey(mol)
if ikey not in vistas:
vistas.add(ikey)
f_out.write(f"{smi},{ikey}\n")
count_unicas += 1
# Notificación de progreso
if count_total % diez_por_ciento == 0:
progreso = (count_total / total_lineas) * 100
print(f"Limpieza: {progreso:.0f}% completado ({count_total} analizadas)...")
sys.stdout.flush()
print("-" * 30)
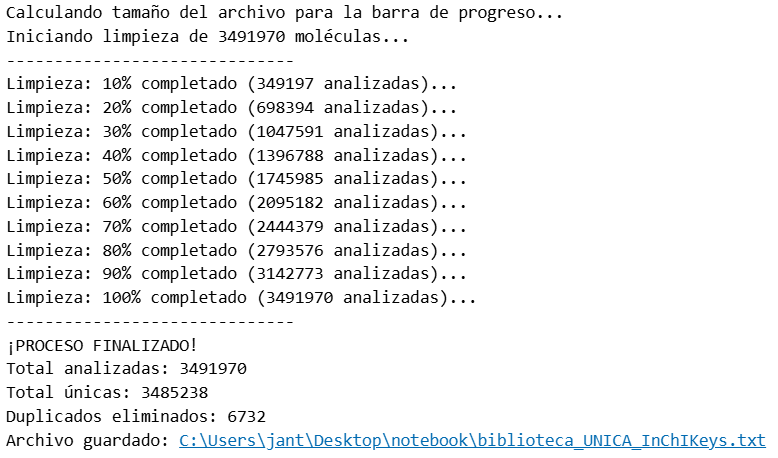
print(f"¡PROCESO FINALIZADO!")
print(f"Total analizadas: {count_total}")
print(f"Total únicas: {count_unicas}")
print(f"Duplicados eliminados: {count_total - count_unicas}")
print(f"Archivo guardado: {output_file}")
# Ejecutar la limpieza
if __name__ == "__main__":
filtrar_duplicados_con_progreso(input_path, output_clean_path)
Con el siguiente script de Python, para ejecutar en Linux, convertiremos los SMILES en moléculas 3D y con H, almacenados en ficheros scaffold-hopping-245_*.sdf con 500 moléculas cada uno.
# -*- coding: utf-8 -*-
import os
import sys
from rdkit import Chem
from rdkit.Chem import AllChem
from concurrent.futures import ProcessPoolExecutor
# --- Configuracion de Rutas ---
INPUT_PATH = '/home/jant.encinar/py-linux/41_quiral-No-quiral/biblioteca_UNICA_InChIKeys.txt'
OUTPUT_DIR = '/home/jant.encinar/py-linux/41_quiral-No-quiral/SDF_outputs'
BASE_NAME = "scaffold-hopping-245"
MOLS_PER_FILE = 500
CPUS = 32 # Debe coincidir con --cpus-per-task en SLURM
def worker_3d(line):
"""
Procesa una linea, genera 3D CON HIDROGENOS y devuelve el bloque SDF como texto.
"""
try:
parts = line.strip().split(',')
if len(parts) < 2:
return None
smiles, ikey = parts[0], parts[1]
mol = Chem.MolFromSmiles(smiles)
if not mol:
return None
# 1. Anadir hidrogenos explicitos antes del 3D
mol = Chem.AddHs(mol)
mol.SetProp("_Name", ikey)
# 2. Asignar estereoquimica
Chem.AssignStereochemistry(mol, force=True, cleanIt=True)
# 3. Generacion de coordenadas 3D para todos los atomos (incluidos H)
params = AllChem.ETKDGv3()
params.randomSeed = 42
if AllChem.EmbedMolecule(mol, params) == 0:
# 4. Optimizacion de energia MMFF94
AllChem.MMFFOptimizeMolecule(mol)
# Devolvemos el bloque SDF completo con coordenadas de hidrogenos
return Chem.MolToMolBlock(mol)
except:
return None
return None
def main():
# Crear directorio si no existe
if not os.path.exists(OUTPUT_DIR):
os.makedirs(OUTPUT_DIR)
print(f"--- Iniciando Procesamiento Paralelo con Hidrogenos (CPUS: {CPUS}) ---")
sys.stdout.flush()
count_success = 0
file_idx = 1
writer = None
with open(INPUT_PATH, 'r') as f_in:
header = f_in.readline() # Saltar cabecera
with ProcessPoolExecutor(max_workers=CPUS) as executor:
# chunksize=100 para optimizar el reparto de tareas en el cluster
results = executor.map(worker_3d, f_in, chunksize=100)
for mol_block in results:
if mol_block:
# Rotacion de archivos cada 500 moleculas
if count_success % MOLS_PER_FILE == 0:
if writer:
writer.close()
file_name = f"{BASE_NAME}_{file_idx:03d}.sdf"
current_sdf_path = os.path.join(OUTPUT_DIR, file_name)
writer = Chem.SDWriter(current_sdf_path)
file_idx += 1
print(f"Escribiendo en: {file_name}")
sys.stdout.flush()
# --- CAMBIO CRITICO: removeHs=False ---
# Esto impide que RDKit borre los H al reconstruir el objeto desde el bloque
tmp_mol = Chem.MolFromMolBlock(mol_block, removeHs=False)
if tmp_mol:
writer.write(tmp_mol)
count_success += 1
# Log de progreso cada 5000 exitos
if count_success % 5000 == 0 and count_success > 0:
print(f"Progreso: {count_success} moleculas guardadas con H en 3D.")
sys.stdout.flush()
if writer:
writer.close()
print(f"--- Proceso Finalizado con exito ---")
print(f"Total moleculas en SDF con hidrogenos: {count_success}")
sys.stdout.flush()
if __name__ == "__main__":
main()
Con el siguiente ejemplo de BASH
#!/bin/bash
#SBATCH --job-name=RDKit_3D # Nombre del trabajo
#SBATCH --output=logs_3d_%j.out # Archivo de salida de logs (%j es el ID del job)
#SBATCH --error=logs_3d_%j.err # Archivo de errores
#SBATCH --nodes=1 # Usamos 1 solo nodo para evitar latencia de red
#SBATCH --ntasks=1 # 1 sola tarea (el script de Python)
#SBATCH --cpus-per-task=32 # IMPORTANTE: Coincidir con 'CPUS = 32' en el script
#SBATCH --mem=64G # RAM de sobra para caché de escritura y objetos Mol
#SBATCH --time=7-0 # Tiempo estimado (24h suele ser suficiente para 32 cores)
#SBATCH --qos=long # Nombre de la partición de tu clúster
# Cargar el entorno de Python/Conda necesario
source /home/jant.encinar/miniconda3/etc/profile.d/conda.sh
conda activate vina # Nombre de mi entorno con RDkit instalado
# Ejecutar el script
python3 /home/jant.encinar/py-linux/41_quiral-No-quiral/53_smiles_combinatoria.py
Script de Cribado ADMET (DILI, hERG, Ames, BBB).
Para implementar DILI, hERG, Ames y BBB de forma gratuita y local con RDKit, usaremos alertas estructurales generadas tras usar diccionarios de fragmentos conocidos por causar toxicidad (como las alertas de Ashby para Ames o fragmentos ionizables para hERG).
Para realizar una selección de compuestos que atraviesen la BBB, no basta con filtrar su LogP (entre 1 y 4), necesitamos también considerar TPSA (Topological Polar Surface Area), que idealmente deberian ser < 60-70 Å2; Pka: estado de ionización (las bases débiles suelen pasar mejor) y HBD (donadores de enlaces de H): idealmente ≥ 3.
El siguiente script de Python filtra atendiendo a los siguientes criterios:
- Prioridad BBB: solo añade al CSV de salida aquellas moléculas que cumplen los criterios fisicoquímicos para cruzar la barrera (baja polaridad, pocos donadores de H y lipofilia moderada).
- Riesgo hERG/DILI: utiliza la "Regla del 2" y la "Regla de Johnson". Se ha demostrado estadísticamente que compuestos con un LogP muy alto y una TPSA muy baja son inhibidores de canales hERG y receptores de toxicidad hepática.
- Ames: utiliza el módulo de fragmentos de RDKit para detectar grupos nitro y azidas, que son "bombas" químicas conocidas por ser mutagénicas.
- SAscore: es un fragmento de código desarrollado originalmente por investigadores de la Universidad de Viena y Novartis, integrado en RDKit, que puntúa del 1 (fácil de sintetizar) al 10 (muy difícil) basándose en la complejidad del esqueleto y los centros quirales.
import pandas as pd
import matplotlib.pyplot as plt
import seaborn as sns
from scipy.stats import mannwhitneyu
import warnings
# Silenciamos avisos innecesarios para que la consola quede limpia
warnings.filterwarnings('ignore', category=FutureWarning)
# --- Configuracion de rutas ---
file_neg = r'D:\encinar\tempjant\108-BBB-prediction\BBB-__RDkit_parameters_distribucion-frecuencias.txt'
file_pos = r'D:\encinar\tempjant\108-BBB-prediction\BBB+__RDkit_parameters_distribucion-frecuencias.txt'
# 1. Cargar y combinar datos
df_neg = pd.read_csv(file_neg, sep='\t')
df_pos = pd.read_csv(file_pos, sep='\t')
df_neg['BBB_Status'] = 'BBB-'
df_pos['BBB_Status'] = 'BBB+'
df_total = pd.concat([df_neg, df_pos], ignore_index=True)
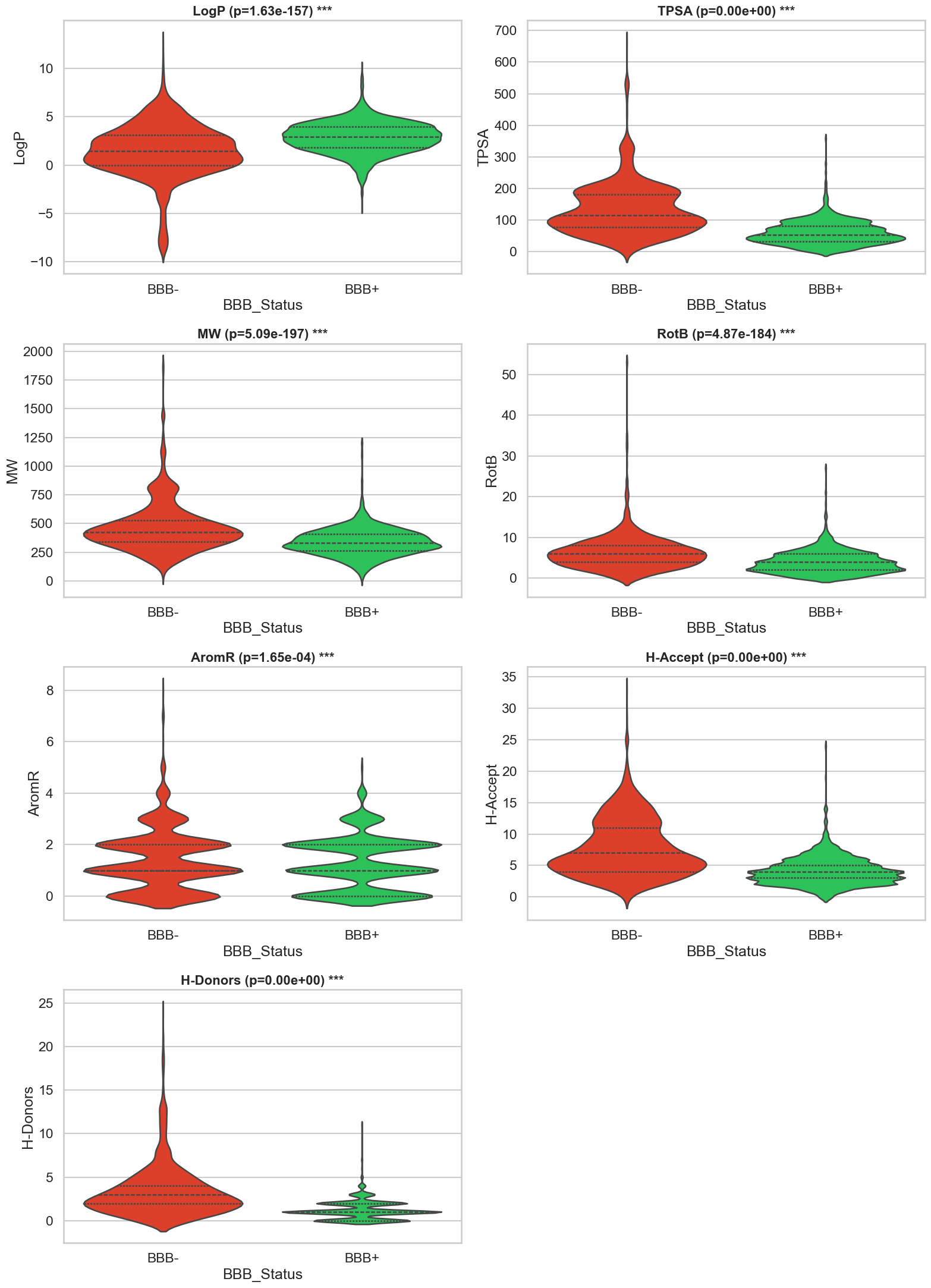
params = ['LogP', 'TPSA', 'MW', 'RotB', 'AromR', 'H-Accept', 'H-Donors']
stats_results = []
# 2. Crear Graficos y calcular Estadisticos
plt.figure(figsize=(16, 22))
sns.set_context("talk")
for i, col in enumerate(params, 1):
# Calcular Test Estadistico
stat, p_val = mannwhitneyu(df_pos[col].dropna(), df_neg[col].dropna())
med_pos = df_pos[col].median()
med_neg = df_neg[col].median()
stats_results.append({'Parametro': col, 'Mediana BBB+': med_pos, 'Mediana BBB-': med_neg, 'p-valor': p_val})
# Grafico de Violin corregido para evitar el Warning
plt.subplot(4, 2, i)
sns.violinplot(
data=df_total,
x='BBB_Status',
y=col,
hue='BBB_Status', # Asignamos hue aqui
palette=["#fa290d", "#13da4f"],
legend=False, # Quitamos la leyenda repetitiva
inner="quart"
)
# Anotacion del p-valor
significancia = "***" if p_val < 0.001 else "*" if p_val < 0.05 else "NS"
plt.title(f'{col} (p={p_val:.2e}) {significancia}', fontsize=16, fontweight='bold')
plt.tight_layout()
plt.savefig(r'D:\encinar\tempjant\108-BBB-prediction\Violin_Plots_BBB_Stats_Clean.png', dpi=300)
# 3. Resumen en consola
df_stats = pd.DataFrame(stats_results)
print("\n--- RESUMEN ESTADISTICO FINAL ---")
print(df_stats.to_string(index=False))
plt.show()
El "perfil farmacóforo ideal" (Basado en B3DB) para un compuesto con alta penetración SNC suele ser:
- TPSA: ≤ 60 o 70 Å2 (la barrera más crítica).
- LogP: Entre 2.0 y 4.0 (lipofilia adecuada).
- H-Donors: ≤ 1 o 2 (los puentes de H "anclan" la molécula en la membrana).
- MW: < 400 (moléculas pequeñas difunden mejor).
- RotB: ≤ 5 (menos entropía conformacional ayuda al paso).
# -*- coding: utf-8 -*-
import os
import sys
import csv
from rdkit import Chem
from rdkit.Chem import Descriptors, rdMolDescriptors, RDConfig
# Import sascorer
try:
sys.path.append(os.path.join(RDConfig.RDContribDir, 'SA_Score'))
import sascorer
except ImportError:
print("Error: sascorer not found.")
sascorer = None
# --- Configuration ---
SDF_DIR = '/home/jant.encinar/py-linux/41_quiral-No-quiral/SDF_outputs/'
OUTPUT_BASE = '/home/jant.encinar/py-linux/41_quiral-No-quiral/LIBRERIA_ULTRA_GOLD_SNC'
OUTPUT_SDF = OUTPUT_BASE + '.sdf'
OUTPUT_CSV = OUTPUT_BASE + '.csv'
def get_ultra_gold_data(mol):
"""Returns a dict with properties if molecule passes, else None"""
if mol is None: return None
# Create clean copy for calculation
clean_mol = Chem.RemoveHs(mol)
# 1. SAscore
sa = 9.9 # Default high if fails
if sascorer:
try:
sa = sascorer.calculateScore(clean_mol)
if sa > 5.0: return None
except: return None
# 2. Physico-chemical
tpsa = Descriptors.TPSA(clean_mol)
logp = Descriptors.MolLogP(clean_mol)
mw = Descriptors.MolWt(clean_mol)
hbd = rdMolDescriptors.CalcNumHBD(clean_mol)
# Veto
if tpsa > 100 or hbd > 4 or mw > 550: return None
# 3. MPO Score
pts = 0
if tpsa <= 75: pts += 1
if 1.5 <= logp <= 4.5: pts += 1
if hbd <= 2: pts += 1
if mw <= 450: pts += 1
if pts >= 3:
# Return data for the CSV
name = mol.GetProp('_Name') if mol.HasProp('_Name') else "NoName"
return {
'ID': name, 'MW': round(mw, 2), 'LogP': round(logp, 2),
'TPSA': round(tpsa, 2), 'HBD': hbd, 'SAscore': round(sa, 2), 'MPO_Points': pts
}
return None
def main():
if not os.path.exists(SDF_DIR):
print(f"Error: Directory {SDF_DIR} not found.")
return
sdf_files = sorted([f for f in os.listdir(SDF_DIR) if f.endswith('.sdf')])
# Setup Output
sdf_writer = Chem.SDWriter(OUTPUT_SDF)
# CSV Setup
csv_file = open(OUTPUT_CSV, 'w', newline='')
fields = ['ID', 'MW', 'LogP', 'TPSA', 'HBD', 'SAscore', 'MPO_Points']
csv_writer = csv.DictWriter(csv_file, fieldnames=fields)
csv_writer.writeheader()
total_in = 0
total_gold = 0
print(f"--- Starting MASSIVE ULTRA GOLD Filter ---")
print(f"Files to process: {len(sdf_files)}")
sys.stdout.flush()
for f in sdf_files:
path = os.path.join(SDF_DIR, f)
suppl = Chem.SDMolSupplier(path, removeHs=False)
for mol in suppl:
if mol is None: continue
total_in += 1
data = get_ultra_gold_data(mol)
if data:
# Write to SDF (Original 3D structure)
sdf_writer.write(mol)
# Write to CSV (Calculated data)
csv_writer.writerow(data)
total_gold += 1
print(f"File {f} | Accumulated Gold: {total_gold}")
sys.stdout.flush()
sdf_writer.close()
csv_file.close()
print(f"\n--- SUCCESS ---")
print(f"Total processed: {total_in}")
print(f"Final Elite Library: {total_gold}")
print(f"Files saved: \n - {OUTPUT_SDF}\n - {OUTPUT_CSV}")
if __name__ == "__main__":
main()
Como nuestro punto limitante es que los nuevos compuestos de interés deben ser BBB+, analizaremos con estos criterios la base de datos B3DB: A curated diverse molecular database of blood-brain barrier permeability with chemical descriptors. Meng F, Xi Y, Huang J, Ayers PW. Scientific Data, 29 Oct 2021, 8(1):289.
1. Información de partida y contexto. El objetivo del script anterior es cribar una librería combinatoria de 1.7 millones de compuestos (en formato SDF 3D con hidrógenos explícitos) para identificar candidatos con alta probabilidad de atravesar la Barrera Hematoencefálica (BHE/BBB) y que, además, presenten una complejidad estructural manejable para su posterior síntesis química.
2. Criterios de selección: el "Perfil MPO-B3DB". A diferencia de las reglas rígidas (como la de Lipinski), este script utiliza un sistema de Optimización Multiparamétrica (MPO) basado en la distribución estadística real de la base de datos B3DB (una de las mayores compilaciones de datos experimentales de permeabilidad cerebral).
Filtros de Veto (Exclusión Absoluta). Se eliminan de entrada compuestos que superan los límites físicos observados en fármacos del SNC, ya que su difusión pasiva es casi nula:
TPSA > 100 Ų: Superficie polar excesiva que impide atravesar lípidos.
HBD > 4: Demasiados donadores de enlaces de hidrógeno "anclan" la molécula a la membrana.
MW > 550 Da: Moléculas demasiado voluminosas para la difusión pasiva eficiente.
Sistema de Puntuación MPO (3/4 para el éxito). Se asigna 1 punto por cada criterio "ideal" cumplido. Una molécula debe sumar al menos 3 puntos para ser considerada Gold:
TPSA ≤ 75 Ų: El "punto dulce" de la polaridad cerebral.
LogP entre 1.5 y 4.5: Balance óptimo entre solubilidad y lipofilia.
HBD ≤ 2: Restricción de donadores (aminas primarias/alcoholes) para favorecer el paso.
MW ≤ 450 Da: Tamaño compacto para mejor permeabilidad.
3. El Filtro de Sintetizabilidad (SAscore). Para evitar "moléculas de ciencia ficción", se integra el algoritmo de Synthetic Accessibility Score (SAscore) de RDKit:
Umbral ≤ 5.0: Tras analizar la base de datos B3DB, observamos que el 75% de los fármacos exitosos tienen un score < 4.4. Situar el corte en 5.0 permite capturar estructuras innovadoras pero químicamente viables, descartando aquellas con excesivos centros quirales o puentes estructurales complejos.
Optimización Técnica: El script realiza una "limpieza de hidrógenos" (RemoveHs) antes del cálculo para evitar que el 3D falsee la complejidad estructural, garantizando resultados realistas.
4. Validación del modelo. Para validar este protocolo, se ejecutó el script sobre el set BBB+ de B3DB (4,956 compuestos que sabemos que entran al cerebro).
Resultados de la validación: rescate: 68.2% (3,383 moléculas). Un rescate de ~70% es un valor de alta confianza.
El 30% restante no capturado suele corresponder a moléculas que utilizan transporte activo (como transportadores de glucosa o aminoácidos), los cuales no se pueden predecir mediante química física simple. Al capturar el 68%, nos aseguramos de que nuestra librería resultante esté compuesta por los mejores candidatos para difusión pasiva, que es el mecanismo más predecible y deseable en el diseño de novo.
Aplicando el mismo script sobre los compuestos que se sabe experimentalmente que son BBB-, rescatamos 756 'falso positivos' de un total de 2849 moléculas. Nos estamos equivocando en un 25%.
5. Salidas del proceso. El script genera:
- SDF Ultra Gold: mantiene las coordenadas 3D y los hidrógenos originales, listo para ser importado directamente en el software de Docking molecular.
- CSV Index: Un inventario detallado con las propiedades físico-químicas calculadas, permitiendo análisis estadísticos instantáneos sin necesidad de re-procesar las moléculas.
La ejecución del script anterior sobre la libreria combinatoria de 3.485.238 compuestos selecciona 14.461, es decir, un 0.4% de moléculas cumplen simultáneamente con un perfil MPO SNC (Probabilidad biológica de cruzar la BBB), SAscore ≤ 5.0 (viabilidad sintética real) ausencia de grupos reactivos o excesivamente polares. El siguiente análisis sobre esta libreria seleccionada nos confirmará si tiene el perfil farmacológico que buscamos o si hay algún sesgo inesperado (por ejemplo, que todas sean muy pequeñas o excesivamente lipofílicas).
import pandas as pd
import matplotlib.pyplot as plt
import seaborn as sns
import os
# --- Configuration ---
csv_path = '/home/jant.encinar/py-linux/41_quiral-No-quiral/LIBRERIA_ULTRA_GOLD_SNC.csv'
output_img = '/home/jant.encinar/py-linux/41_quiral-No-quiral/PERFIL_ELITE_SNC.png'
# Load data
df = pd.read_csv(csv_path)
# Setup plotting style
sns.set_theme(style="whitegrid")
fig, axes = plt.subplots(2, 2, figsize=(16, 12))
fig.suptitle(f'Análisis de la Librería Élite SNC (n = {len(df)})', fontsize=20, fontweight='bold')
# 1. TPSA vs LogP (The "CNS Sweet Spot")
# Standard CNS drugs usually cluster: TPSA < 75 and LogP 1-4
sns.kdeplot(ax=axes[0,0], data=df, x='TPSA', y='LogP', fill=True, cmap='rocket', thresh=0, levels=15)
axes[0,0].set_title('Mapa de Densidad: TPSA vs LogP', fontsize=14)
axes[0,0].axhline(4.5, color='black', linestyle='--', alpha=0.5)
axes[0,0].axvline(75, color='black', linestyle='--', alpha=0.5)
axes[0,0].set_xlabel('TPSA (Ų)')
# 2. Molecular Weight vs SAscore (Complexity vs Size)
hb = axes[0,1].hexbin(df['MW'], df['SAscore'], gridsize=25, cmap='YlGnBu', mincnt=1)
fig.colorbar(hb, ax=axes[0,1], label='Frecuencia')
axes[0,1].set_title('Viabilidad Sintética vs Tamaño Molecular', fontsize=14)
axes[0,1].set_xlabel('Peso Molecular (Da)')
axes[0,1].set_ylabel('SAscore (1=Fácil, 5=Límite)')
# 3. Distribution of MPO Points
sns.countplot(ax=axes[1,0], data=df, x='MPO_Points', palette='viridis')
axes[1,0].set_title('Calidad del Perfil MPO', fontsize=14)
axes[1,0].set_xlabel('Puntos acumulados (3-4)')
axes[1,0].set_ylabel('Número de Moléculas')
# 4. Boxplot of Properties (Summary)
props = df[['LogP', 'SAscore', 'HBD']]
sns.boxplot(ax=axes[1,1], data=props, palette='Set2')
axes[1,1].set_title('Distribución de Parámetros Clave', fontsize=14)
plt.tight_layout(rect=[0, 0.03, 1, 0.95])
plt.savefig(output_img, dpi=300)
print(f"--- Gráficos generados correctamente en: {output_img} ---")
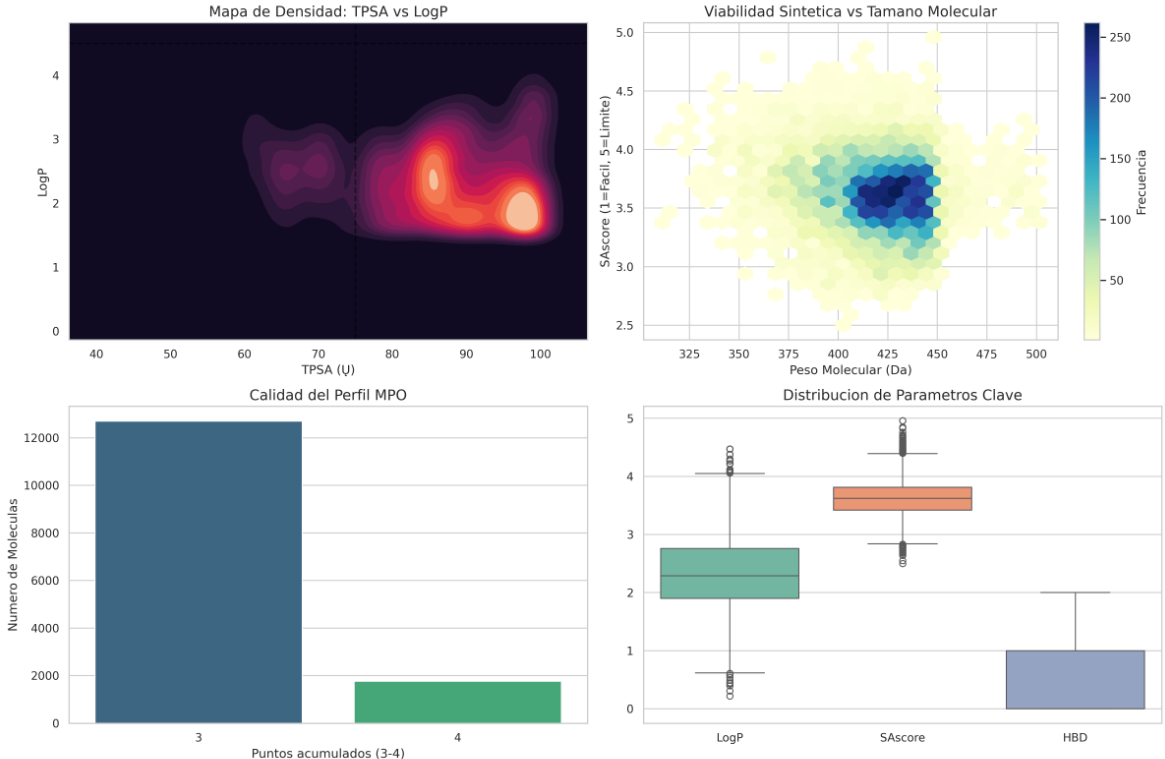
El "Mapa de Calor" (TPSA vs LogP).
- hay una población muy fuerte alrededor de TPSA 85-95 y LogP 2,0-2,5.
- hay otra población en la zona de TPSA 100 y LogP 1,5.
- aunque muchas moléculas están cerca del límite superior de TPSA (100), se mantienen en un rango de LogP muy equilibrado. Esto sugiere que son moléculas con una polaridad moderada pero no excesiva.
Viabilidad Sintética vs Tamaño (MW vs SAscore).
- La mancha azul oscura (la mayoría de tus 14.461 moléculas) está centrada en MW 425 Da y SAscore 3,6.
- Un SAscore de 3,6 es muy bueno; son moléculas con una complejidad estructural interesante (probablemente varios anillos o centros quirales) pero totalmente accesibles para síntesis. Hemos evitado la zona "peligrosa" (SAscore > 4.5).
Calidad del Perfil MPO.
- Hay unas ~12.500 moléculas con 3 puntos y unas ~1.800 moléculas con 4 puntos (el perfil perfecto).
- Esos 1.800 compuestos con 4 puntos son candidatos "VIPs". Podemos priorizar a estas 1.800 en el siguiente paso de docking sobre la diana proteíca.
Distribución de parámetros clave (Boxplot).
- El LogP tiene una mediana muy sana (~2.3), lo que augura buena solubilidad.
- El SAscore es muy compacto, lo que indica que el filtro de sintetizabilidad funcionó de forma muy uniforme.
- El HBD (donadores de Hidrógeno) está mayoritariamente en 0 o 1, lo cual favorece la entrada por la barrera hematoencefálica. Pocos donadores = menos resistencia al cruce.
Vamos a concentrar la potencia de cálculo (1000 rounds por ligando) en los 1.800 candidatos "perfectos" usando GPU. Estas moléculas no sólo cumplen con la farmacofonía del SNC, sino que tienen el balance físico-químico ideal.
El siguiente script realiza la selección sobre el archivo /home/jant.encinar/py-linux/41_quiral-No-quiral/LIBRERIA_ULTRA_GOLD_SNC.sdf.
# -*- coding: utf-8 -*-
import os
from rdkit import Chem
from rdkit.Chem import Descriptors, rdMolDescriptors
# --- Rutas de archivos ---
SDF_INPUT = '/home/jant.encinar/py-linux/41_quiral-No-quiral/LIBRERIA_ULTRA_GOLD_SNC.sdf'
SDF_OUTPUT = '/home/jant.encinar/py-linux/41_quiral-No-quiral/ELITE_MPO4_1800.sdf'
def es_mpo4(mol):
if mol is None: return False
# Trabajamos sobre una copia sin Hidrogenos para los descriptores
clean_mol = Chem.RemoveHs(mol)
tpsa = Descriptors.TPSA(clean_mol)
logp = Descriptors.MolLogP(clean_mol)
mw = Descriptors.MolWt(clean_mol)
hbd = rdMolDescriptors.CalcNumHBD(clean_mol)
# Contador de puntos MPO (Criterios estrictos de la B3DB)
points = 0
if tpsa <= 75: points += 1
if 1.5 <= logp <= 4.5: points += 1
if hbd <= 2: points += 1
if mw <= 450: points += 1
return points == 4
def main():
if not os.path.exists(SDF_INPUT):
print(f"Error: No se encuentra el archivo {SDF_INPUT}")
return
suppl = Chem.SDMolSupplier(SDF_INPUT, removeHs=False) # Mantenemos 3D e Hidrogenos
writer = Chem.SDWriter(SDF_OUTPUT)
total_analizadas = 0
total_extraidas = 0
print("--- Iniciando extraccion de moleculas MPO=4 ---")
for mol in suppl:
if mol is None: continue
total_analizadas += 1
if es_mpo4(mol):
writer.write(mol)
total_extraidas += 1
if total_analizadas % 1000 == 0:
print(f"Procesadas: {total_analizadas} | Extraidas: {total_extraidas}")
writer.close()
print(f"\n--- EXTRACCION COMPLETADA ---")
print(f"Moleculas analizadas: {total_analizadas}")
print(f"Moleculas MPO=4 guardadas: {total_extraidas}")
print(f"Archivo listo para Docking GPU: {SDF_OUTPUT}")
if __name__ == "__main__":
main()
El siguiente script de Python analiza cuantos ficheros *.sdf se encuentren en la ruta D:\aaa y genera en la ruta D:\bbb ficheros de ULTRA_GOLD_SNC-850-*.sdf con 500 moléculas. Listos para ser usados en simulaciones de molecular docking.
import os
import glob
from datetime import datetime
# Rutas
input_dir = r"D:\aaa"
output_dir = r"D:\bbb"
# Parametros
molecules_per_file = 500
output_prefix = "ULTRA_GOLD_SNC-850-"
os.makedirs(output_dir, exist_ok=True)
sdf_files = sorted(glob.glob(os.path.join(input_dir, "*.sdf")))
file_index = 1
molecule_count_in_file = 0
total_molecules = 0
out_file = None
current_output_filename = None
def open_new_output_file(index):
global current_output_filename
filename = f"{output_prefix}{index:04d}_ligands.sdf"
path = os.path.join(output_dir, filename)
current_output_filename = filename
print(f"[{datetime.now().strftime('%H:%M:%S')}] CREANDO fichero de salida: {filename}")
return open(path, "w", encoding="utf-8")
out_file = open_new_output_file(file_index)
current_molecule_lines = []
for sdf_file in sdf_files:
print(f"Procesando fichero de entrada: {os.path.basename(sdf_file)}")
with open(sdf_file, "r", encoding="utf-8", errors="ignore") as f:
for line in f:
current_molecule_lines.append(line)
if line.strip() == "$$$$":
out_file.writelines(current_molecule_lines)
current_molecule_lines.clear()
molecule_count_in_file += 1
total_molecules += 1
if molecule_count_in_file == molecules_per_file:
out_file.close()
print(
f"[{datetime.now().strftime('%H:%M:%S')}] "
f"CERRADO {current_output_filename} "
f"({molecule_count_in_file} moléculas, "
f"total acumulado: {total_molecules})"
)
file_index += 1
molecule_count_in_file = 0
out_file = open_new_output_file(file_index)
# Cierre del ultimo fichero
if out_file:
out_file.close()
print(
f"[{datetime.now().strftime('%H:%M:%S')}] "
f"CERRADO {current_output_filename} "
f"({molecule_count_in_file} moléculas, "
f"total acumulado: {total_molecules})"
)
print(f"Proceso finalizado correctamente. Moleculas totales procesadas: {total_molecules}")