MolPort database v8.
El siguiente script de Python lee todos los ficheros SDF que hemos descargado desde /FTP_PROFESSIONAL/2026-03/All Stock Compounds en nuestra ruta E:\MolPort-database-v8 y genera en la ruta E:\MolPort-database-v8_SDF500 ficheros SDF con 500 moléculas filtradas usando los criterios siguientes: 1) que exista una sola molécula (elimina fragmentos, sales, iones, etc), 2) peso molecular entre 250 y 700 Da, 3) compuestos sin carbonos quirales, 4) extrae el nombre de MolPort del campo "PUBCHEM_EXT_DATASOURCE_REGID" y lo coloca en cada molecula seleccionada, 5) genera moléculas 3D y añade Hidrógenos.
Al final de esta página se describe como crear un entorno desde "conda (base)", llamado molport-db necesario para ejecutar los scripts contenidos en esta página.
import os
import glob
import multiprocessing
from io import BytesIO
from rdkit import Chem
from rdkit.Chem import AllChem, Descriptors
from tqdm import tqdm
from pebble import ProcessPool
from concurrent.futures import TimeoutError
# ==========================================
# FUNCIÓN DE TRABAJO (Se ejecuta en paralelo)
# ==========================================
def process_single_block(mol_block_str):
supplier = Chem.ForwardSDMolSupplier(BytesIO(mol_block_str.encode('utf-8')))
try:
mol = next(supplier)
except StopIteration:
mol = None
if mol is None:
return ("error_lectura", None, None)
# 1. Criterio: Una sola molécula
if len(Chem.GetMolFrags(mol)) > 1:
return ("multi_frag", None, None)
# 2. Criterios: Peso molecular entre 250 y 700 Da
mw = Descriptors.MolWt(mol)
if mw > 700.0:
return ("peso_alto", None, None)
if mw < 250.0:
return ("peso_bajo", None, None)
# 3. Criterio: Sin carbonos quirales
chiral_centers = Chem.FindMolChiralCenters(mol, includeUnassigned=True)
for idx, _ in chiral_centers:
if mol.GetAtomWithIdx(idx).GetAtomicNum() == 6:
return ("quirales", None, None)
# 4. Extraer el nombre de MolPort
if mol.HasProp("PUBCHEM_EXT_DATASOURCE_REGID"):
mol_name = mol.GetProp("PUBCHEM_EXT_DATASOURCE_REGID")
else:
mol_name = "Unknown_MolPort_ID"
# 5. Generar 3D y añadir Hidrógenos
mol_with_h = Chem.AddHs(mol)
res = AllChem.EmbedMolecule(mol_with_h, randomSeed=42, maxAttempts=1000)
if res == -1:
return ("error_3d", None, None)
# IMPORTANTE: Devolvemos 3 cosas (Estado, Molécula en binario, Nombre extraído)
return ("ok", mol_with_h.ToBinary(), mol_name)
# ==========================================
# LECTOR DE TEXTO
# ==========================================
def sdf_block_generator(filepath):
with open(filepath, 'r', encoding='utf-8', errors='ignore') as f:
block = []
for line in f:
block.append(line)
if line.strip() == "$$$$":
yield "".join(block)
block = []
# ==========================================
# FUNCIÓN PRINCIPAL
# ==========================================
def process_molport_database():
input_dir = r"E:\MolPort-database-v8"
output_dir = r"E:\MolPort-database-v8_SDF500"
os.makedirs(output_dir, exist_ok=True)
sdf_files = glob.glob(os.path.join(input_dir, "*.sdf"))
if not sdf_files:
print(f"No se encontraron archivos .sdf en {input_dir}")
return
mols_per_file = 500
file_counter = 1
current_mol_count = 0
stats = {
"leidas": 0, "guardadas": 0, "descartadas_multi_frag": 0,
"descartadas_peso_alto": 0, "descartadas_peso_bajo": 0,
"descartadas_quirales": 0, "descartadas_error_3d": 0,
"descartadas_timeout": 0, "errores_lectura": 0
}
current_out_filepath = os.path.join(output_dir, f"MPv8-{file_counter:04d}.sdf")
writer = Chem.SDWriter(current_out_filepath)
cpu_cores = multiprocessing.cpu_count()
print(f"Iniciando. CPU: {cpu_cores} núcleos. Timeout: 30s. Límites PM: 250-700 Da.")
with ProcessPool(max_workers=cpu_cores) as pool:
for filepath in sdf_files:
filename = os.path.basename(filepath)
blocks_iterator = sdf_block_generator(filepath)
future = pool.map(process_single_block, blocks_iterator, timeout=30, chunksize=10)
iterator = future.result()
with tqdm(desc=f"Procesando {filename}") as pbar:
while True:
try:
# Ahora desempaquetamos las 3 variables
status, mol_bin, mol_name = next(iterator)
stats["leidas"] += 1
pbar.update(1)
if status == "ok":
mol = Chem.Mol(mol_bin)
# RE-ASIGNAMOS EL NOMBRE justo antes de guardar
mol.SetProp("_Name", mol_name)
writer.write(mol)
current_mol_count += 1
stats["guardadas"] += 1
if current_mol_count == mols_per_file:
writer.close()
file_counter += 1
current_mol_count = 0
current_out_filepath = os.path.join(output_dir, f"MPv8-{file_counter:04d}.sdf")
writer = Chem.SDWriter(current_out_filepath)
else:
if status == "error_lectura":
stats["errores_lectura"] += 1
else:
stats[f"descartadas_{status}"] += 1
except StopIteration:
break
except TimeoutError:
stats["leidas"] += 1
stats["descartadas_timeout"] += 1
pbar.update(1)
except Exception as error:
stats["leidas"] += 1
stats["errores_lectura"] += 1
pbar.update(1)
if writer is not None:
writer.close()
if current_mol_count == 0 and os.path.exists(current_out_filepath):
os.remove(current_out_filepath)
print("\n" + "="*50)
print("RESUMEN DEL PROCESAMIENTO")
print("="*50)
print(f"Total de moléculas leídas: {stats['leidas']}")
print(f"Moléculas guardadas con éxito: {stats['guardadas']}")
print("-" * 50)
print("Descartadas:")
print(f" - Por múltiples fragmentos: {stats['descartadas_multi_frag']}")
print(f" - Por PM > 700 Da: {stats['descartadas_peso_alto']}")
print(f" - Por PM < 250 Da: {stats['descartadas_peso_bajo']}")
print(f" - Por tener carbonos quirales: {stats['descartadas_quirales']}")
print(f" - Por fallo al generar el 3D: {stats['descartadas_error_3d']}")
print(f" - Por exceder tiempo (30s): {stats['descartadas_timeout']}")
print(f" - Por error de lectura RDKit: {stats['errores_lectura']}")
print("="*50)
if __name__ == "__main__":
process_molport_database()
Después de unas 10 horas de cálculo en una máquina Windows, usando 99% de CPU (8 cores) y unos 8 GB de RAM generamos 6391 ficheros SDF.
==================================================
RESUMEN DEL PROCESAMIENTO
==================================================
Total de moléculas leídas: 5940456
Moléculas guardadas con éxito: 3195634
--------------------------------------------------
Descartadas:
- Por múltiples fragmentos: 322548
- Por PM > 700 Da: 24197
- Por PM < 250 Da: 492999
- Por tener carbonos quirales: 1904378
- Por fallo al generar el 3D: 206
- Por exceder tiempo (30s): 106
- Por error de lectura RDKit: 388
==================================================El siguiente script de Python nos permite calcular unos parámetros de ADMET de la libreria completa. Cuestion técnica importante: para sacar todo el partido a la programación multitarea de este y del anterior script, ambos deben ejecutarse desde una consola de 'conda' como administrador y en un entorno activo que tenga instalado 'RDkit', 'pandas', 'tqdm', etc.
import os
import glob
import multiprocessing
import pandas as pd
import matplotlib.pyplot as plt
import seaborn as sns
from rdkit import Chem
from rdkit.Chem import Descriptors, QED
from tqdm import tqdm
# ==========================================
# CONFIGURACIÓN DE TOXICÓFOROS (SMARTS)
# ==========================================
# Alertas estructurales simplificadas para detectar posibles riesgos
TOX_ALERTS = {
"Ames": [Chem.MolFromSmarts(s) for s in ['c[N+](=O)[O-]', 'c[NH2]', 'C1OC1']], # Nitroaromáticos, aminas aromáticas, epóxidos
"hERG": [Chem.MolFromSmarts(s) for s in ['[NX3+]', '[NX3;!$(NC=O)]']], # Aminas básicas fuertemente asociadas a bloqueo hERG
"DILI": [Chem.MolFromSmarts(s) for s in ['[cX3](=O)[OX2H1]', '[NX3](=O)=O', 'C(=O)C(=O)']] # Alertas hepáticas genéricas
}
# ==========================================
# FUNCIONES DE CÁLCULO
# ==========================================
def calculate_logs_esol(mol, logp, mw, nrot):
"""Calcula LogS usando la ecuación ESOL (Delaney)"""
aromatic_atoms = sum(1 for atom in mol.GetAtoms() if atom.GetIsAromatic())
heavy_atoms = mol.GetNumHeavyAtoms()
ap = aromatic_atoms / heavy_atoms if heavy_atoms > 0 else 0
# Ecuación ESOL
return 0.16 - 0.63 * logp - 0.0062 * mw + 0.066 * nrot - 0.74 * ap
def check_tox(mol, alert_list):
"""Verifica si la molécula contiene algún toxicóforo de la lista"""
for pattern in alert_list:
if pattern is not None and mol.HasSubstructMatch(pattern):
return "Positivo"
return "Negativo"
# ==========================================
# PROCESAMIENTO DE UN ARCHIVO SDF
# ==========================================
def process_sdf_file(filepath):
"""Lee un archivo SDF entero y calcula ADMET para todas sus moléculas"""
results = []
supplier = Chem.SDMolSupplier(filepath, removeHs=False) # Mantenemos los H que ya añadiste
for mol in supplier:
if mol is None:
continue
try:
# Identificación
mol_name = mol.GetProp("_Name") if mol.HasProp("_Name") else "Unknown"
# Descriptores 1D y 2D
mw = Descriptors.MolWt(mol)
natom = mol.GetNumHeavyAtoms() # Usamos átomos pesados (sin contar H)
nhet = Descriptors.NumHeteroatoms(mol)
nring = Descriptors.RingCount(mol)
nrot = Descriptors.NumRotatableBonds(mol)
hba = Descriptors.NumHAcceptors(mol)
hbd = Descriptors.NumHDonors(mol)
tpsa = Descriptors.TPSA(mol)
logp = Descriptors.MolLogP(mol)
qed = QED.qed(mol)
# LogS
logs = calculate_logs_esol(mol, logp, mw, nrot)
# --- Clasificación BBB ---
# NOTA: Has pedido HBD >= 3. Si quisieras aplicar la regla estándar de química médica,
# cambia el "hbd >= 3" por "hbd <= 3".
if (1 <= logp <= 4) and (tpsa <= 70) and (hbd <= 3):
bbb_status = "BBB+"
else:
bbb_status = "BBB-"
# Toxicidad (Alertas estructurales)
ames_status = check_tox(mol, TOX_ALERTS["Ames"])
herg_status = check_tox(mol, TOX_ALERTS["hERG"])
dili_status = check_tox(mol, TOX_ALERTS["DILI"])
# Añadir a la lista de resultados
results.append({
"ID": mol_name,
"MW": round(mw, 2),
"nAtom": natom,
"nHet": nhet,
"nRing": nring,
"nRot": nrot,
"HBA": hba,
"HBD": hbd,
"TPSA": round(tpsa, 2),
"logP": round(logp, 2),
"logS": round(logs, 2),
"QED": round(qed, 3),
"BBB": bbb_status,
"Ames": ames_status,
"hERG": herg_status,
"DILI": dili_status
})
except Exception as e:
continue # Si falla una molécula por cualquier motivo matemático, la saltamos
return results
# ==========================================
# FUNCIÓN PRINCIPAL Y GRAFICADO
# ==========================================
def main():
input_dir = r"E:\aaa"
output_file = os.path.join(input_dir, "_MPv8-ADMET.txt")
sdf_files = glob.glob(os.path.join(input_dir, "*.sdf"))
if not sdf_files:
print("No se encontraron archivos SDF.")
return
print(f"Calculando descriptores ADMET para {len(sdf_files)} archivos...")
print("Usando multiprocesamiento. Esto irá muy rápido.")
all_results = []
cpu_cores = multiprocessing.cpu_count()
# Procesamiento en paralelo de los archivos
with multiprocessing.Pool(processes=cpu_cores) as pool:
# Usamos imap_unordered para que la barra de progreso se actualice rápido
for res_batch in tqdm(pool.imap_unordered(process_sdf_file, sdf_files), total=len(sdf_files), desc="Archivos procesados"):
all_results.extend(res_batch)
# Guardar resultados en un DataFrame de Pandas
print(f"\nGuardando {len(all_results)} resultados en: {output_file}")
df = pd.DataFrame(all_results)
# Guardar como archivo de texto separado por tabuladores (TXT/TSV)
df.to_csv(output_file, sep="\t", index=False)
print("Generando gráficos de distribución...")
# --- CREACIÓN DE LOS GRÁFICOS (Seaborn/Matplotlib) ---
sns.set_theme(style="whitegrid")
# Variables continuas que queremos dibujar en histogramas
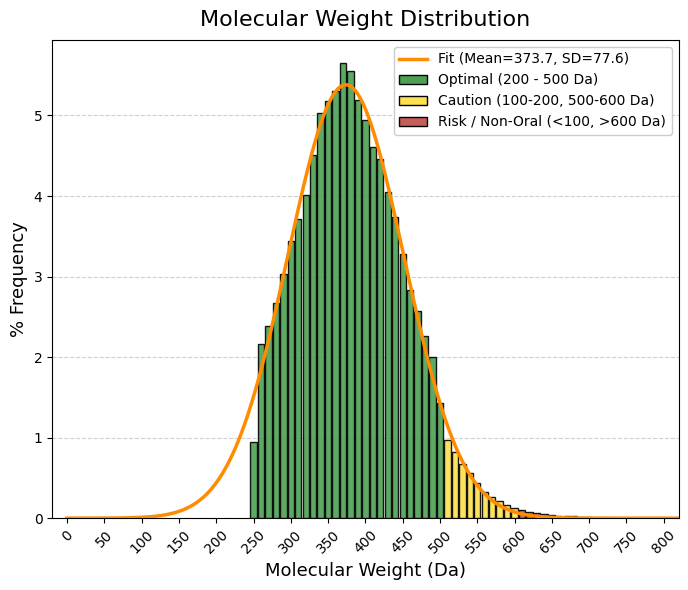
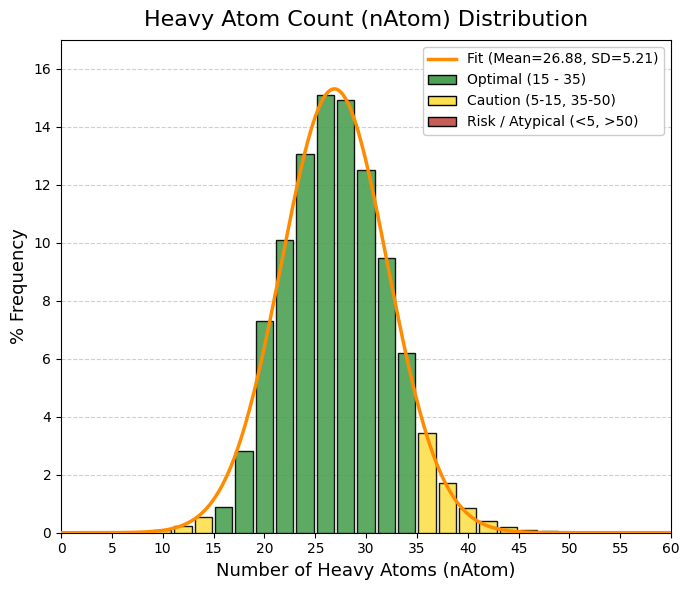
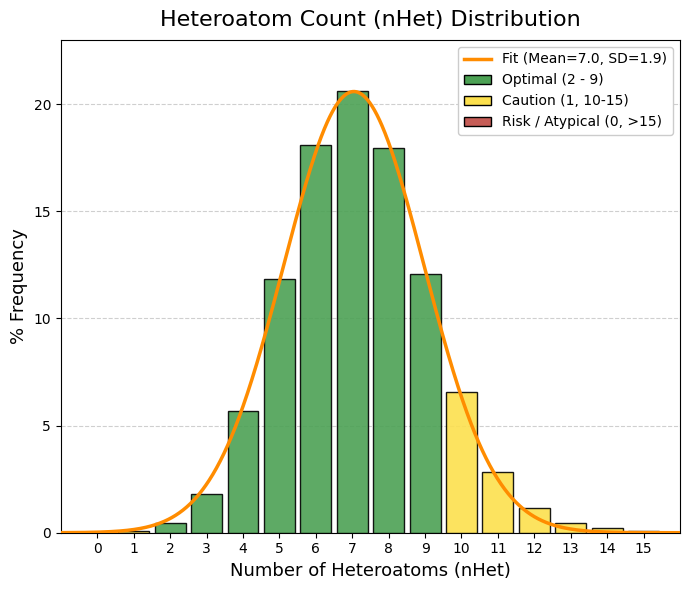
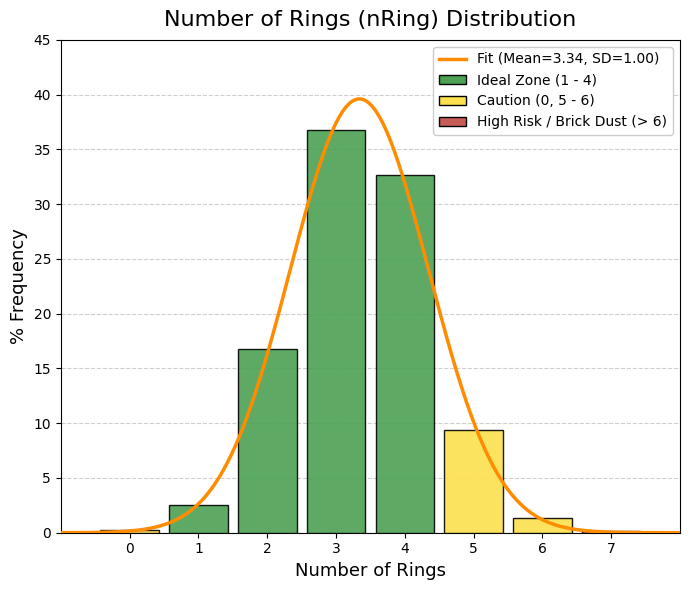
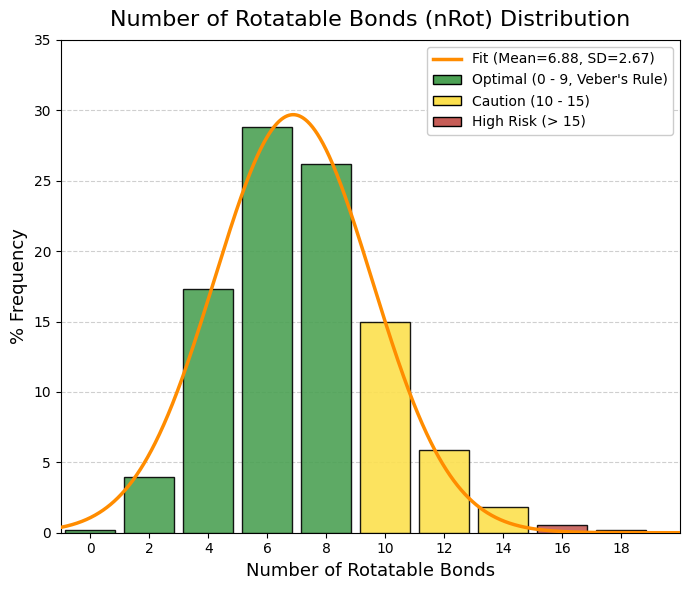
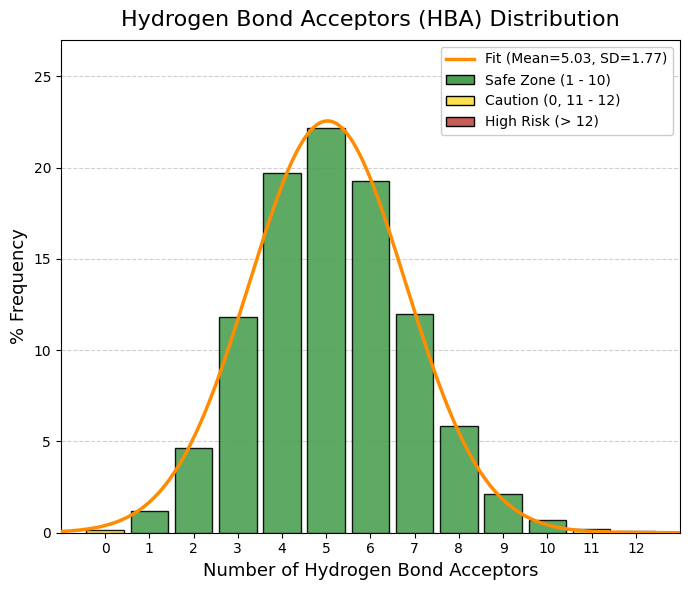
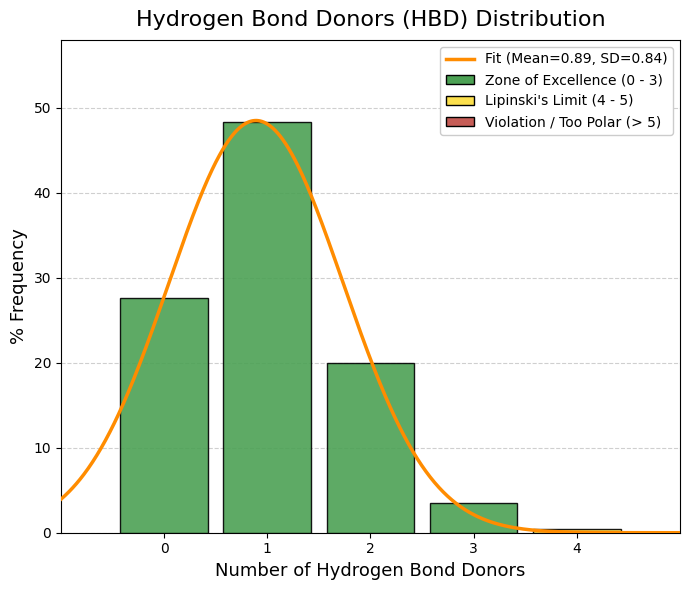
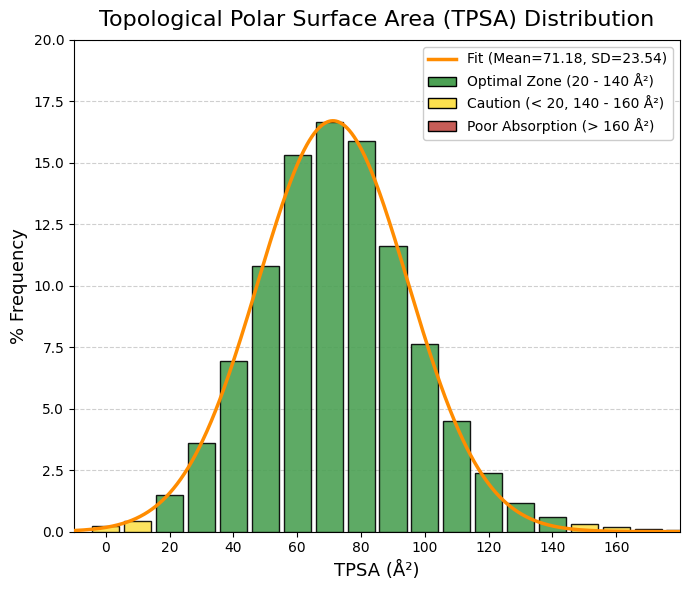
vars_to_plot = ["MW", "TPSA", "logP", "logS", "QED", "nRot", "nRing", "HBA", "HBD"]
fig, axes = plt.subplots(3, 3, figsize=(15, 12))
fig.suptitle('Distribución de Parámetros Físico-Químicos de la Librería MolPort', fontsize=16)
axes = axes.flatten()
for i, var in enumerate(vars_to_plot):
sns.histplot(df[var], bins=40, ax=axes[i], kde=True, color="steelblue")
axes[i].set_title(f"Distribución de {var}")
axes[i].set_ylabel("Frecuencia")
axes[i].set_xlabel(var)
plt.tight_layout(rect=[0, 0.03, 1, 0.95])
# Imprimir un pequeño resumen por consola antes de mostrar la gráfica
print("\n" + "="*40)
print("RESUMEN DE CLASIFICACIONES BINARIAS")
print("="*40)
print(df['BBB'].value_counts().to_string())
print("-" * 20)
print(df['Ames'].value_counts().to_string())
print("-" * 20)
print(df['hERG'].value_counts().to_string())
print("-" * 20)
print(df['DILI'].value_counts().to_string())
print("="*40)
# Mostrar la gráfica interactiva en VS Code
plt.show()
if __name__ == "__main__":
main()
 |
 |
 |
 |
 |
 |
 |
 |
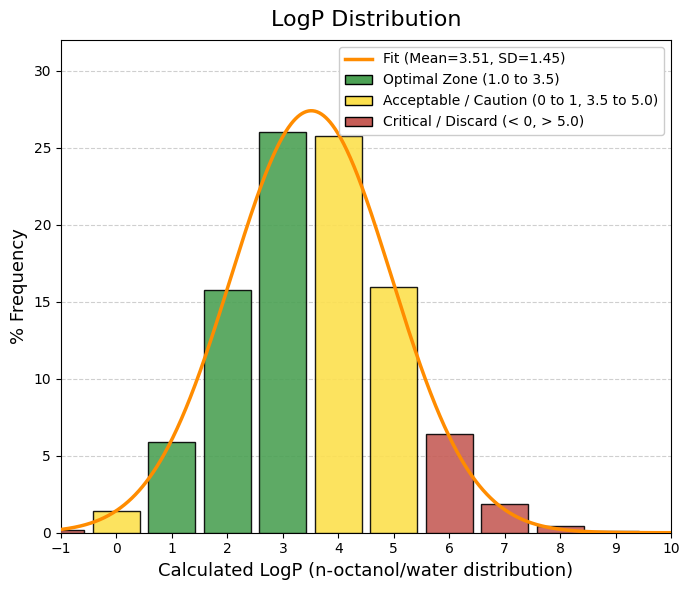 |
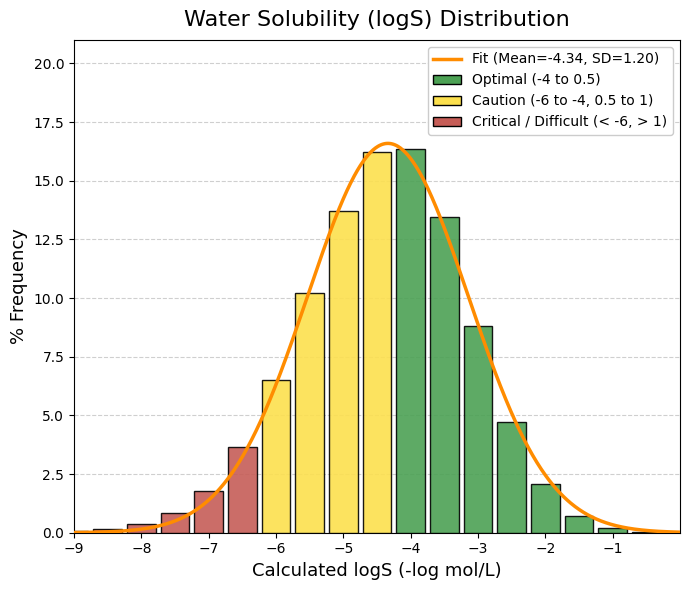 |
Creación del entorno 'molport-db' desde 'conda (base)' para ejecutar los scripts de Python anteriores.
1. Crear el entorno y activar
Primero, asegúrate de estar en tu terminal (base) y crea el entorno con una versión estable de Python (la 3.10 o 3.11 suelen ser ideales para estas librerías).
conda create -n molport-db python=3.10 -y
conda activate molport-db2. Instalación de librerías
Para optimizar la compatibilidad, lo ideal es instalar lo más pesado (RDKit y herramientas científicas) vía Conda (o el canal conda-forge) y dejar las utilidades específicas para Pip.
conda install -c conda-forge rdkit pandas matplotlib seaborn tqdm -yAlgunas librerías como pebble (para el manejo avanzado de procesos) no siempre están disponibles o actualizadas en los canales principales de Conda, por lo que las instalamos con Pip:
pip install pebbleTambién podemos usar la versión 'todo-en-uno'.
conda create -n molport-db python=3.10 -y && conda activate molport-db && conda install -c conda-forge rdkit pandas matplotlib seaborn tqdm -y && pip install pebbleFiltrado por precio...
El siguiente script de Python usa los ficheros *.sdf descargados desde el FTP de MolPort para seleccionar el MolPort_ID de los compuestos que en el campo PRICERANGE_1MG tengan un valor < 50.
import os
import glob
import time
def extract_cheap_molecules():
# 1. Definir rutas
input_dir = r"E:\MolPort-database-v8"
output_filepath = r"E:\MolPort-database-v8\MPv8____PRICERANGE_1MG___menos-50-USD.txt"
# Buscar todos los archivos .sdf en el directorio
sdf_files = glob.glob(os.path.join(input_dir, "*.sdf"))
if not sdf_files:
print(f"No se encontraron archivos .sdf en: {input_dir}")
return
print(f"Se han encontrado {len(sdf_files)} archivos SDF. Iniciando extracción...")
start_time = time.time()
# Contadores para las estadísticas finales
total_molecules = 0
saved_molecules = 0
# 2. Abrir el archivo de salida en modo escritura
with open(output_filepath, 'w', encoding='utf-8') as outfile:
# Escribir la cabecera del archivo (opcional pero recomendable)
outfile.write("PUBCHEM_EXT_DATASOURCE_REGID\tPRICERANGE_1MG\n")
# 3. Iterar sobre cada archivo SDF
for filepath in sdf_files:
filename = os.path.basename(filepath)
print(f"Procesando: {filename}...")
# Variables temporales para la molécula actual
current_regid = None
current_price = None
# Flags para saber qué estamos leyendo
reading_regid = False
reading_price = False
# Leer el SDF línea a línea para no colapsar la memoria RAM
with open(filepath, 'r', encoding='utf-8') as infile:
for line in infile:
line = line.strip()
# Identificador de fin de molécula en un SDF
if line == "$$$$":
total_molecules += 1
# Si tenemos ambos datos y el precio es el que buscamos, lo guardamos
if current_regid and current_price == "< 50":
outfile.write(f"{current_regid}\t{current_price}\n")
saved_molecules += 1
# Resetear variables para la siguiente molécula
current_regid = None
current_price = None
reading_regid = False
reading_price = False
continue
# Detectar los campos de datos (Data Headers)
if line.startswith("> ") or line.startswith("> "):
reading_regid = True
continue
elif line.startswith("> ") or line.startswith("> "):
reading_price = True
continue
# Capturar los valores en la línea inmediatamente inferior al Header
if reading_regid:
if line: # Ignorar líneas vacías
current_regid = line
reading_regid = False
elif reading_price:
if line:
current_price = line
reading_price = False
# 4. Resumen final
elapsed_time = time.time() - start_time
print("-" * 50)
print("¡Proceso completado con éxito!")
print(f"Total de moléculas evaluadas: {total_molecules}")
print(f"Moléculas guardadas (< 50 USD): {saved_molecules}")
print(f"Archivo guardado en: {output_filepath}")
print(f"Tiempo total: {elapsed_time:.2f} segundos.")
if __name__ == "__main__":
extract_cheap_molecules()
# ¡Proceso completado con éxito!
# Total de moléculas evaluadas: 5941410
# Moléculas guardadas (< 50 USD): 2019242
# Archivo guardado en: E:\MolPort-database-v8\MPv8____PRICERANGE_1MG___menos-50-USD.txt
# Tiempo total: 664.55 segundos.
Con el siguiente script de Python rescataremos del total de moleculas 3D con H las que satisfagan la condición de valor menor de 50 USD en el campo PRICERANGE_1MG.
import os
import glob
import time
def extract_3d_molecules_simple_sdf():
# 1. Definir rutas exactas
txt_filepath = r"E:\MolPort-database-v8\MPv8____PRICERANGE_1MG___menos-50-USD.txt"
input_dir = r"E:\MolPort-database-v8_SDF500"
output_dir = r"E:\MolPort-database-v8_SDF500__menos-50-USD"
# Crear directorio de salida si no existe
if not os.path.exists(output_dir):
os.makedirs(output_dir)
print("Paso 1: Cargando la lista de IDs a buscar...")
start_time = time.time()
target_ids = set()
with open(txt_filepath, 'r', encoding='utf-8') as f:
# Leer línea a línea separando por el tabulador
for line in f:
parts = line.strip().split('\t')
# Evitar meter la cabecera si la tiene
if parts and not parts[0].startswith("PUBCHEM"):
target_ids.add(parts[0])
print(f"¡Cargados {len(target_ids)} IDs únicos en {(time.time() - start_time):.2f} segundos!")
# 2. Buscar todos los archivos SDF en subcarpetas
print("\nPaso 2: Buscando archivos .sdf en las subcarpetas...")
# os.walk es extremadamente robusto para navegar por estructuras complejas de carpetas en Windows
sdf_files = []
for root, dirs, files in os.walk(input_dir):
for file in files:
if file.endswith(".sdf"):
sdf_files.append(os.path.join(root, file))
print(f"Se han encontrado {len(sdf_files)} archivos SDF para procesar.")
# 3. Variables de control para la generación de lotes
file_counter = 1
mol_counter_in_file = 0
total_saved = 0
# Función auxiliar para crear el nombre de archivo
def get_output_filename(counter):
return os.path.join(output_dir, f"MPv8-50USD-{counter:04d}_ligands.sdf")
print("\nPaso 3: Extrayendo moléculas y generando lotes...")
current_out_file = open(get_output_filename(file_counter), 'w', encoding='utf-8')
# 4. Procesar cada archivo encontrado
for filepath in sdf_files:
with open(filepath, 'r', encoding='utf-8') as infile:
mol_lines = []
current_id = None
for line in infile:
# Si la lista de líneas está vacía, estamos en la primera línea de la molécula
if not mol_lines:
current_id = line.strip()
mol_lines.append(line)
# Fin de la molécula
if line.strip() == "$$$$":
# Comprobar si el ID de la primera línea está en nuestro Set
if current_id in target_ids:
current_out_file.writelines(mol_lines)
mol_counter_in_file += 1
total_saved += 1
# Si llegamos a 500, cerramos y abrimos el siguiente archivo
if mol_counter_in_file == 500:
current_out_file.close()
file_counter += 1
current_out_file = open(get_output_filename(file_counter), 'w', encoding='utf-8')
mol_counter_in_file = 0
# Resetear para la siguiente molécula
mol_lines = []
current_id = None
# 5. Cierre y limpieza final
current_out_file.close()
# Si el último archivo se quedó vacío (porque cuadró justo múltiplo de 500), lo borramos
if mol_counter_in_file == 0:
os.remove(get_output_filename(file_counter))
file_counter -= 1
elapsed_time = time.time() - start_time
print("-" * 50)
print("¡Proceso completado con éxito!")
print(f"Total de moléculas extraídas y guardadas: {total_saved}")
print(f"Total de archivos SDF generados: {file_counter}")
print(f"Directorio de salida: {output_dir}")
print(f"Tiempo total: {elapsed_time:.2f} segundos.")
if __name__ == "__main__":
extract_3d_molecules_simple_sdf()
MolPort v8, no Quiral, precio de 1MG < 50 USD, 250 y 700 Da tiene un total de 1.285.720 moléculas.
MolPort v8, Quiral, precio de 1MG < 50 USD, 250 y 700 Da tiene un total de 974.350 moléculas.
Con el siguiente script de Python vamos a rescatar de la libreria inicial /FTP_PROFESSIONAL/2026-03/All Stock Compounds los compuestos con un máximo de 2 carbonos quirales, precio de 1MG < 50 USD, 250 y 700 Da. Eso sí, vamos a ejecutarlo en Linux, con 32 CPUs y 64 GB de RAM.
import os
import glob
import time
import multiprocessing as mp
from rdkit import Chem
from rdkit.Chem import AllChem
from rdkit.Chem.EnumerateStereoisomers import EnumerateStereoisomers, StereoEnumerationOptions
from rdkit import RDLogger
# --- 1. PATH DEFINITIONS (Edit these paths for your Linux server) ---
INPUT_DIR = "/home/jant/runDC"
OUTPUT_DIR = "/home/jant/runMD"
CPUS_A_USAR = 32
# -------------------------------------------------------------------
def procesar_archivo(filepath):
"""
This function will be executed in parallel by each CPU core.
It processes an entire SDF file.
"""
# Disable warnings in each child process to avoid console spam
RDLogger.DisableLog('rdApp.*')
filename = os.path.basename(filepath)
base_name = os.path.splitext(filename)[0]
supplier = Chem.ForwardSDMolSupplier(filepath, sanitize=True)
stereo_opts = StereoEnumerationOptions(tryEmbedding=True, maxIsomers=4)
file_counter = 1
mol_counter_in_file = 0
isomers_saved = 0
mols_evaluated = 0
def get_new_writer(counter):
out_path = os.path.join(OUTPUT_DIR, f"MPv8-Q_{base_name}_{counter:04d}.sdf")
return Chem.SDWriter(out_path)
writer = get_new_writer(file_counter)
for mol in supplier:
mols_evaluated += 1
if mol is None:
continue
try:
# --- FILTERS ---
if not mol.HasProp("PRICERANGE_1MG"): continue
if "< 50" not in mol.GetProp("PRICERANGE_1MG"): continue
if not mol.HasProp("PUBCHEM_EXT_DATASOURCE_REGID"): continue
base_id = mol.GetProp("PUBCHEM_EXT_DATASOURCE_REGID")
if len(Chem.GetMolFrags(mol)) > 1: continue
chiral_centers = Chem.FindMolChiralCenters(mol, includeUnassigned=True)
num_chiral = len(chiral_centers)
if num_chiral < 1 or num_chiral > 2: continue
# --- 3D GENERATION AND STEREOISOMERS ---
isomers = list(EnumerateStereoisomers(mol, options=stereo_opts))
for idx, isomer in enumerate(isomers):
isomer_name = f"{base_id}_{idx+1}"
isomer_h = Chem.AddHs(isomer)
res = AllChem.EmbedMolecule(isomer_h, randomSeed=42)
if res != 0:
res = AllChem.EmbedMolecule(isomer_h, useRandomCoords=True, randomSeed=42)
if res != 0: continue
try:
AllChem.MMFFOptimizeMolecule(isomer_h, maxIters=200)
except:
pass
# Calculate R/S labels
try:
Chem.AssignStereochemistry(isomer_h, force=True, cleanIt=True)
centros_rs = Chem.FindMolChiralCenters(isomer_h)
if centros_rs:
texto_quiral = " | ".join([f"C{i}: {rs}" for i, rs in centros_rs])
isomer_h.SetProp("QUIRALIDAD", texto_quiral)
else:
isomer_h.SetProp("QUIRALIDAD", "No defined centers")
except:
pass
isomer_h.SetProp("_Name", isomer_name)
# --- THE SOLUTION TO THE KEKULIZE ERROR ---
try:
writer.write(isomer_h)
mol_counter_in_file += 1
isomers_saved += 1
except Exception:
# If it fails due to KekulizeException or any other reason, silently ignore
continue
# ----------------------------------------
if mol_counter_in_file == 500:
writer.close()
file_counter += 1
writer = get_new_writer(file_counter)
mol_counter_in_file = 0
except Exception:
# If a molecule is totally corrupted, catch the error and move to the next
continue
if writer is not None:
writer.close()
if mol_counter_in_file == 0:
last_file = os.path.join(OUTPUT_DIR, f"MPv8-Q_{base_name}_{file_counter:04d}.sdf")
if os.path.exists(last_file):
os.remove(last_file)
return mols_evaluated, isomers_saved, filename
def main():
if not os.path.exists(OUTPUT_DIR):
os.makedirs(OUTPUT_DIR)
print("Starting search in multiprocess mode (32 CPUs)...")
start_time = time.time()
sdf_files = glob.glob(os.path.join(INPUT_DIR, "*.sdf"))
if not sdf_files:
print(f"No files found in: {INPUT_DIR}")
return
print(f"Files found: {len(sdf_files)}")
print("Background processing has started. Please wait...\n")
total_mols = 0
total_isomers = 0
# Create a Pool with the specified number of CPUs
with mp.Pool(CPUS_A_USAR) as pool:
for result in pool.imap_unordered(procesar_archivo, sdf_files):
mols_eval, iso_saved, fname = result
total_mols += mols_eval
total_isomers += iso_saved
print(f"File {fname} completed. (Added {iso_saved} isomers)")
elapsed_time = time.time() - start_time
print("-" * 50)
print("PROCESS COMPLETED SUCCESSFULLY IN MULTIPROCESSING!")
print(f"Initial molecules evaluated: {total_mols}")
print(f"Stereoisomers (3D+H) saved: {total_isomers}")
print(f"Total time: {elapsed_time/60:.2f} minutes.")
if __name__ == '__main__':
main()
# Justo debajo tenemos el sbatch. Esto no es parte del script...
#!/bin/bash
#SBATCH --job-name=MP8-Q
#SBATCH --qos=normal
#SBATCH --cpus-per-task=32
#SBATCH --mem=64G
#SBATCH --time=7-0
#SBATCH --output=/home/jant/py-linux/02-MP8-Quiral/MP8-Q_salida-%j.out
#SBATCH --error=/home/jant/py-linux/02-MP8-Quiral/MP8-Q_error-%j.err
# Cargar el entorno de Python/Conda necesario
source /home/jant/anaconda3/etc/profile.d/conda.sh
# Nombre de mi entorno con RDkit instalado
conda activate vina125
# Ejecutar el script
python3 /home/jant/py-linux/02-MP8-Quiral/extractor_quirales_3d_linux.py
File iis-005-000-000--005-499-999.sdf completed. (Added 6938 isomers)
File iis-005-500-000--005-999-999.sdf completed. (Added 9643 isomers)
File iis-003-000-000--003-499-999.sdf completed. (Added 29170 isomers)
File iis-004-500-000--004-999-999.sdf completed. (Added 22408 isomers)
File iis-002-500-000--002-999-999.sdf completed. (Added 62150 isomers)
File iis-003-500-000--003-999-999.sdf completed. (Added 48609 isomers)
File iis-002-000-000--002-499-999.sdf completed. (Added 69800 isomers)
File iis-004-000-000--004-499-999.sdf completed. (Added 75298 isomers)
File iis-001-500-000--001-999-999.sdf completed. (Added 91966 isomers)
File iis-000-500-000--000-999-999.sdf completed. (Added 148804 isomers)
File iis-001-000-000--001-499-999.sdf completed. (Added 195120 isomers)
File iis-000-000-000--000-499-999.sdf completed. (Added 214444 isomers)
--------------------------------------------------
PROCESS COMPLETED SUCCESSFULLY IN MULTIPROCESSING!
Initial molecules evaluated: 5941410
Stereoisomers (3D+H) saved: 974350
Total time: 881.09 minutes.