Predicción de homodímeros en AlphaFold 3
La predicción de un homodímero en AlphaFold3 es posible a través de un entorno web gratuito y muy intuitivo llamado AlphaFold Server, identificado con un usuario de Google.
Una vez dentro de la interfaz de envío de trabajos, en el cuadro de texto de esa Entity 1, pega la secuencia de aminoácidos en formato texto limpio (solo las letras, sin espacios ni saltos de línea).
Justo al de la secuencia, verás un campo llamado 'Copies', al ser un homodímero, ambas cadenas son exactamente iguales. Por lo tanto, simplemente cambia el número de copias de 1 a 2.
¿Cómo evaluar si el modelo de AlphaFold3 es bueno?
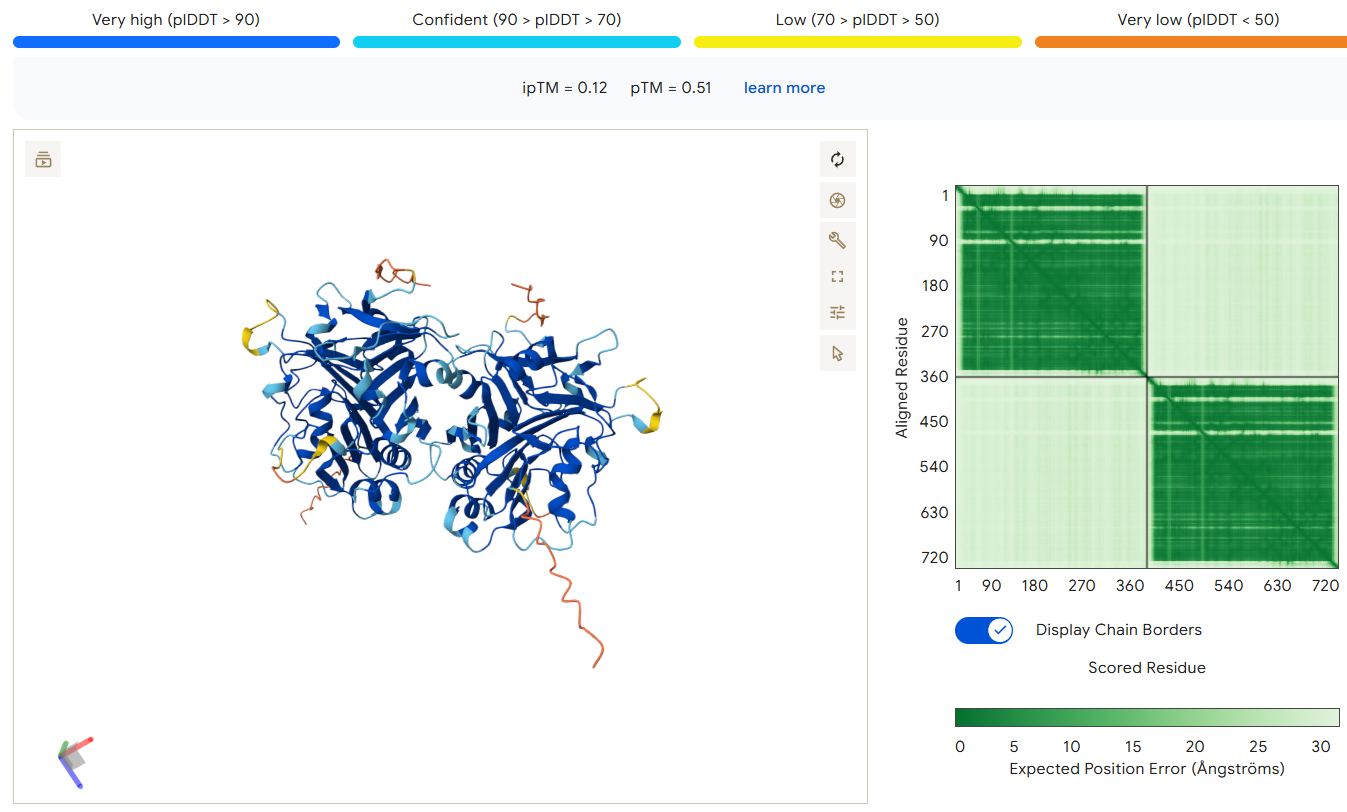
Cuando termine, el servidor te mostrará un visor 3D y varias métricas de calidad que equivalen a los Z-Scores de SWISS-MODEL. Debes fijarte en:
ipTM (Interface pTM): es la métrica más importante y mide la confianza de la interacción en la interfaz donde se unen las dos cadenas.
- > 0.85: Excelente interacción y alta confianza.
- 0.70 - 0.85: Buena predicción de la interfaz.
- < 0.50: AlphaFold ha juntado las proteínas al azar o la interacción propuesta no es fiable.
pLDDT (confianza por residuo): te pintará el modelo en azul (muy confiable, >90), amarillo o naranja (baja confianza, <50, suelen ser regiones intrínsecamente desordenadas o loops).
En el ejemplo siguiente, se muestra un modelo con una baja ipTM: 0.12, por tanto, la interacción propuesta no es fiable, sobre todo sabiendo que "we demonstrate that recombinant Burkholderia thailandensis DNase II is highly active at low pH in the absence of divalent metal ions, similar to eukaryotic DNase II. The crystal structure of B. thailandensis DNase II shows a dimeric quaternary structure which appears capable of binding double-stranded DNA" (DOI: 10.1093/nar/gkx222).
En vista a este fracaso, es importante considerar otras estrategias para obtener una predicción más precisa de la estructura del homodímero. Hemos usado 5I3E como molde para generar la predicción en SWISS-MODEL.
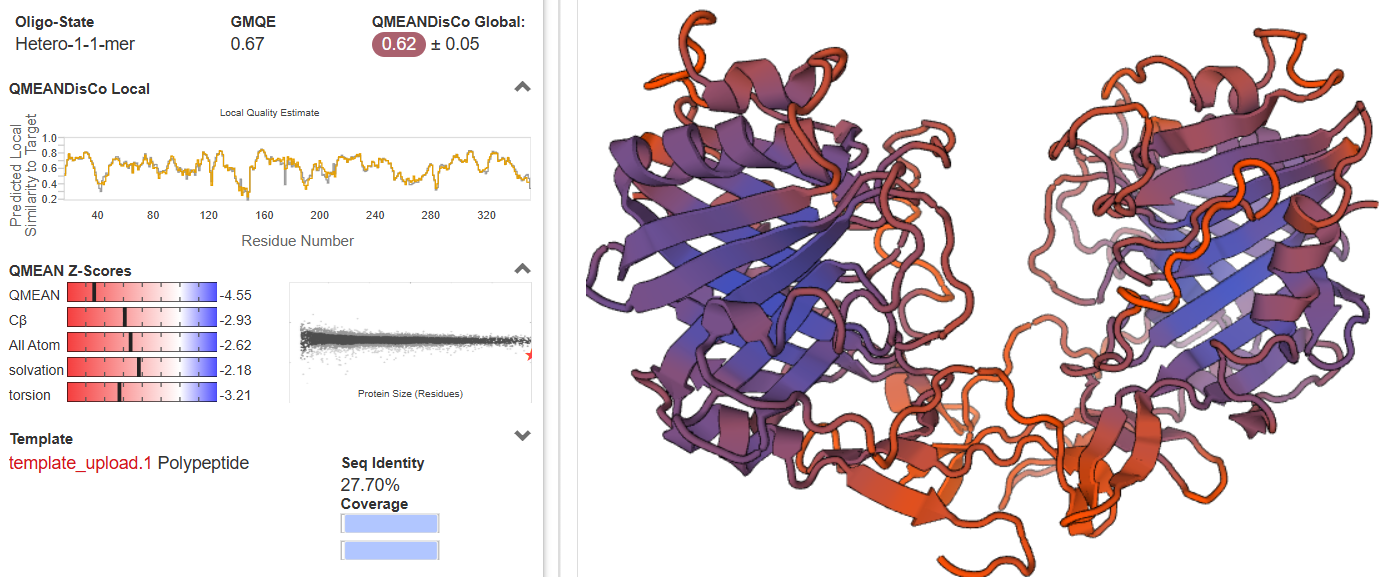
A partir de los datos que se muestran de SWISS-MODEL, el modelo que obtenido se puede clasificar como regular tirando a malo (o aceptable con reservas).
Para calificarlo de esta manera, nos basamos en los tres parámetros clave de validación estructural que aparecen en la imagen:
1. Identidad de Secuencia (Seq Identity): 27.70% entre la secuencia del modelo (O00115) y la secuencia del molde (Q2T8B0).
En el modelado por homología, el "límite de la zona de penumbra" se sitúa alrededor del 30%. Con un 27.70%, estamos justo en el límite o ligeramente por debajo. Esto significa que, aunque el plegamiento del esqueleto polipeptídico probablemente sea el correcto, los detalles de las cadenas laterales, los 'loops' y las interacciones específicas pueden contener bastantes errores.
2. GMQE (Global Model Quality Estimation): 0.67
GMQE es una estimación global de la calidad del modelo que combina las propiedades de la alineación de secuencias y la estructura de la plantilla. Varía entre 0 y 1. Un valor de 0.67 es un número moderado. Indica que el modelo tiene una fiabilidad aceptable en su conjunto para representar la estructura general, pero no es una predicción de alta precisión (los modelos muy buenos suelen superar el 0.80).
3. QMEANDisCo Global: 0.62 ± 0.05 y QMEAN Z-Scores
El Z-Score compara el modelo con estructuras reales de alta resolución resueltas por cristalografía de rayos X. Un valor cercano a 0 (zona blanca) significa que tu modelo tiene una calidad similar a una estructura real experimental. Valores negativos (hacia el rojo) indican que el modelo es de peor calidad. Todos los indicadores (QMEAN, Cβ, All Atom, solvation, torsion) están en la zona roja, con valores que van desde -2.18 hasta -4.55. Z-Score inferior a -2.0 (y especialmente por debajo de -3.0 como en la torsión o el QMEAN general) es una señal de alerta. Esto está diciendo que la geometría local de los aminoácidos, las fuerzas de solvatación y las distorsiones de los ángulos de torsión son significativamente peores o menos "naturales" que las de una proteína real promedio.
De la plantilla 5I3E, basándonos en este reporte oficial de validación del Protein Data Bank, diríamos que es una estructura experimental de una calidad excepcionalmente alta. Si el modelo obtenido antes con SWISS-MODEL era regular tirando a malo (o aceptable con reservas), el problema no era de la plantilla. 5I3E es un molde casi perfecto. La razón de que sus métricas sean tan buenas se lee directamente en los gráficos de percentiles, donde cuanto más a la derecha (zona azul) esté la marca, mejor es la calidad:
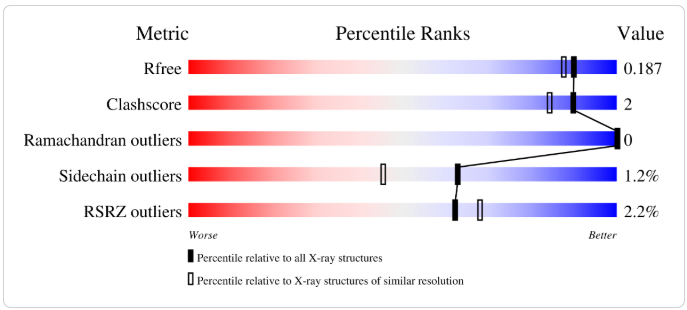
1. Rfree: 0.187
Rfree mide la concordancia entre el modelo atómico teórico y los datos experimentales de difracción de rayos X. Un valor de 0.187 es magnífico para una estructura de rayos X. Significa que los datos experimentales respaldan con mucha precisión la posición real de los átomos. Está firmemente asentado en la zona azul (alta calidad).
2. Clashscore: 2
Clashscore cuenta el número de choques estéreos (dos átomos no enlazados queintentan ocupar el mismo espacio) por cada 1000 átomos. Un valor de 2 es extremadamente bajo. Indica que prácticamente no hay solapamientos atómicos anómalos. La estructura está físicamente muy bien relajada y optimizada.
3. Ramachandran outliers: 0
Evalúa si los ángulos de torsión del esqueleto de la proteína (phi y psi) son geométricamente estables y naturales. Un valor de 0 significa que ningún aminoácido está en una conformación forzada o energéticamente desfavorable.
4. Sidechain outliers: 1.2%
Mide el porcentaje de cadenas laterales que adoptan rotámeros (conformaciones espaciales) inusuales o poco probables. Solo un 1.2% está fuera de lo común. Es un porcentaje bajísimo, lo que supone que los aminoácidos individuales están modelados con un realismo impecable.
5. RSRZ outliers: 2.2%
Mide la cantidad de aminoácidos cuyo ajuste local con el mapa de densidad electrónica es deficiente (zonas borrosas en el experimento). Un 2.2% es un valor completamente normal y bajo, habitualmente restringido a los extremos (extremo N o C-terminal) o a bucles muy flexibles expuestos al solvente que se mueven de forma natural.