Calcula grid-center y dimensiones de caja para hacer simulaciones de docking con vina-1.2.5
Puedes descargar vina 1.2.5., tanto para windows como para Linux. Guarda el siguiente script de Python para ejecutar en Windows como "docking_box_with_drawing.py". Crea una escena de Pymol y separa cada ligando en un objeto. Desde la consola de PyMol ejecuta como "run docking_box_with_drawing.py"
# El siguiente ejemplo se ha calculado sobre 5JJR.pdb,
# posición del ligando SAH y calcula el centro del ligando y una caja con \
# dimensiones de 4 A más a sus dimensiones máximas.
PyMOL>run docking_box_with_drawing.py
PyMOL>calculate_docking_box("SAH", expansion=4.0)
center_x = 27.373, center_y = 129.089, center_z = 18.808
size_x = 13.501, size_y = 23.419, size_z = 12.406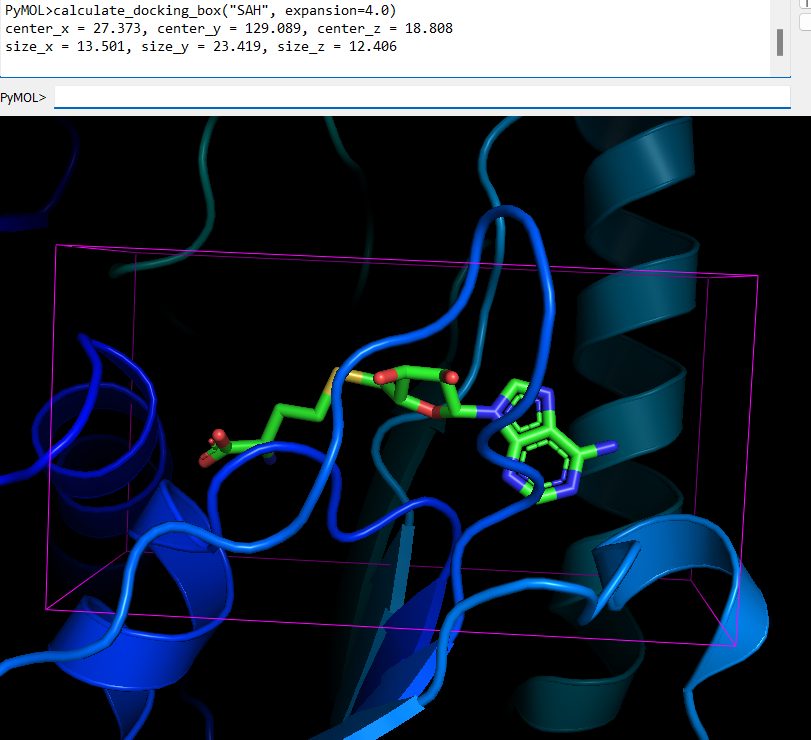
from pymol import cmd, cgo
def calculate_docking_box(obj_name, expansion=7.0):
"""
Calcula el centro y tamaño de la caja para docking molecular y la dibuja en PyMOL.
Args:
obj_name (str): Nombre del objeto en la escena de PyMOL.
expansion (float): Expansión en Ångströms para calcular el tamaño de la caja.
Ejemplo de uso:
calculate_docking_box("68E", expansion=7.0)
Returns:
None: Imprime los valores del centro y tamaño en consola y dibuja la caja.
"""
# Obtener las coordenadas mínimas y máximas del objeto
min_coord, max_coord = cmd.get_extent(obj_name)
# Calcular el centro
center_x = (min_coord[0] + max_coord[0]) / 2.0
center_y = (min_coord[1] + max_coord[1]) / 2.0
center_z = (min_coord[2] + max_coord[2]) / 2.0
# Calcular el tamaño con expansión
size_x = (max_coord[0] - min_coord[0]) + 2 * expansion
size_y = (max_coord[1] - min_coord[1]) + 2 * expansion
size_z = (max_coord[2] - min_coord[2]) + 2 * expansion
# Imprimir los valores en consola
print(f"center_x = {center_x:.3f}, center_y = {center_y:.3f}, center_z = {center_z:.3f}")
print(f"size_x = {size_x:.3f}, size_y = {size_y:.3f}, size_z = {size_z:.3f}")
# Dibujar la caja en la escena
draw_box(center_x, center_y, center_z, size_x, size_y, size_z)
def draw_box(center_x, center_y, center_z, size_x, size_y, size_z):
"""
Dibuja una caja en la escena de PyMOL usando las coordenadas del centro y dimensiones.
Args:
center_x, center_y, center_z (float): Coordenadas del centro.
size_x, size_y, size_z (float): Dimensiones de la caja.
"""
half_size_x = size_x / 2.0
half_size_y = size_y / 2.0
half_size_z = size_z / 2.0
# Calcular las esquinas de la caja
corners = [
[center_x - half_size_x, center_y - half_size_y, center_z - half_size_z],
[center_x + half_size_x, center_y - half_size_y, center_z - half_size_z],
[center_x + half_size_x, center_y + half_size_y, center_z - half_size_z],
[center_x - half_size_x, center_y + half_size_y, center_z - half_size_z],
[center_x - half_size_x, center_y - half_size_y, center_z + half_size_z],
[center_x + half_size_x, center_y - half_size_y, center_z + half_size_z],
[center_x + half_size_x, center_y + half_size_y, center_z + half_size_z],
[center_x - half_size_x, center_y + half_size_y, center_z + half_size_z],
]
# Crear las líneas de la caja (enlazar las esquinas)
box_cgo = [
cgo.BEGIN, cgo.LINES,
cgo.LINEWIDTH, 20.0, # Grosor de las líneas
# cgo.COLOR, 1.0, 1.0, 1.0 # Color blanco (RGB)
cgo.COLOR, 1.0, 0.0, 1.0 # Color magenta (RGB)
]
edges = [
(0, 1), (1, 2), (2, 3), (3, 0), # Parte inferior
(4, 5), (5, 6), (6, 7), (7, 4), # Parte superior
(0, 4), (1, 5), (2, 6), (3, 7) # Conexiones verticales
]
for start, end in edges:
box_cgo.extend([
cgo.VERTEX, *corners[start],
cgo.VERTEX, *corners[end]
])
box_cgo.append(cgo.END)
# Dibujar la caja en PyMOL
cmd.load_cgo(box_cgo, "docking_box")Puedes usar YASARA para guardar el fichero del receptor 5JJR.pdbqt, y del ligando lig-68E.pdbqt o lig-SAH.pdbqt y construir estos ficheros a partir de los SMILES
# Usa YASARA
SavePDBQTR 1 ,D:\autodock-vina\5JJR.pdbqt,transform=Yes #receptor 5JJR
SavePDBQT 1 ,D:\autodock-vina\lig-68E.pdbqt,transform=Yes #ligand 68E
SavePDBQT 1 ,D:\autodock-vina\lig-SAH.pdbqt,transform=Yes #ligand SAH
BuildSMILES String="CC1=CC(=C(C=C1C(=O)NS(=O)(=O)C2=CC=CC3=C2N=CC=C3)C4=CC=C(S4)C#CCO)OC"
BuildSMILES String="N[CH](CCSC[CH]1O[CH]([CH](O)[CH]1O)n2cnc3c(N)ncnc23)C(O)=O" #SAH
# Usa el script de Python arriba escrito:
calculate_docking_box("68E", expansion=4.0)
center_x = 27.523, center_y = 175.912, center_z = 18.436
size_x = 15.581, size_y = 24.891, size_z = 12.990
calculate_docking_box("SAH", expansion=4.0)
center_x = 27.373, center_y = 129.089, center_z = 18.808
size_x = 13.501, size_y = 23.419, size_z = 12.406
# Usa el desde CMD las siguientes sentencias:
vina_1.2.5_win.exe --receptor 5JJR.pdbqt --ligand lig-68E.pdbqt --center_x 27.523 --center_y 175.912
--center_z 18.436 --size_x 15.581 --size_y 24.891 --size_z 12.990 --exhaustiveness 32 --out
5JJR_lig-68E_complex.pdbqt --num_modes=10 --spacing 0.375 --scoring vina --cpu 8 >5JJR_lig-68E_complex.txt
vina_1.2.5_win.exe --receptor 5JJR.pdbqt --ligand lig-SAH.pdbqt --center_x 27.523 --center_y 129.089
--center_z 18.808 --size_x 13.501 --size_y 23.419 --size_z 12.406 --exhaustiveness 32 --out
5JJR_lig-SAH_complex.pdbqt --num_modes=10 --spacing 0.375 --scoring vina --cpu 8 >5JJR_lig-SAH_complex.txtA continuación vamos a usar la libreria generada en un post anterior para hacer simulaciones de docking usando vina 1.2.5. Esta libreria tiene ficheros SDF de 3000 moléculas. El primer paso necesario es convertir cada molécula en un fichero PDBQT. a continuación se muestran los script para Windows y para Linux. Las rutas deben adecuarse a nuestras necesidades y debemos tener instalados módulos de Python como RDkit, numpy, pandas, openbabel, etc, deseablemente en un entorno virtual, que es muy fácil de crear con Miniconda.
Versión para ejecutar en Windows.
import os
from rdkit import Chem
from rdkit.Chem import AllChem
import subprocess
import re
def sdf_to_pdbqt_with_hydrogens(sdf_file, output_folder, vina_exe, receptor_pdbqt, vina_output_folder, deltaG_file):
"""
Convierte un archivo SDF a múltiples archivos PDBQT, agrega hidrógenos explícitos, y ejecuta Vina para cada uno de los ficheros PDBQT generados.
Además, guarda un registro de los archivos procesados y extrae la afinidad (delta G) del archivo de salida de Vina.
Args:
sdf_file (str): Ruta al archivo SDF de entrada.
output_folder (str): Carpeta de salida para los archivos PDBQT.
vina_exe (str): Ruta al ejecutable de Vina.
receptor_pdbqt (str): Ruta al archivo PDBQT del receptor.
vina_output_folder (str): Carpeta de salida para los resultados de Vina.
deltaG_file (str): Ruta al archivo de salida donde se guardarán los resultados de la afinidad.
"""
# Crear la carpeta de salida si no existe
os.makedirs(output_folder, exist_ok=True)
os.makedirs(vina_output_folder, exist_ok=True)
# Leer moléculas del archivo SDF
supplier = Chem.SDMolSupplier(sdf_file)
# Crear o cargar el archivo de registro de archivos procesados
processed_file = os.path.join(output_folder, "processed_files.txt")
if os.path.exists(processed_file):
with open(processed_file, "r") as f:
processed_files = set(f.read().splitlines())
else:
processed_files = set()
# Verificar si podemos abrir el archivo deltaG.txt para escritura
try:
with open(deltaG_file, "a") as f:
pass
except Exception as e:
print(f"Error al intentar abrir el archivo {deltaG_file} para escritura: {e}")
return
for i, mol in enumerate(supplier):
if mol is None:
print(f"Molécula {i+1} no válida, saltando...")
continue
# Obtener el nombre de la molécula
mol_name = mol.GetProp("_Name") if mol.HasProp("_Name") else f"mol_{i+1}"
# Comprobar si el archivo PDBQT ya ha sido procesado
pdbqt_file = os.path.join(output_folder, f"{mol_name}.pdbqt")
if pdbqt_file in processed_files:
print(f"El archivo {pdbqt_file} ya ha sido procesado, saltando...")
continue
# Agregar hidrógenos explícitos
mol = Chem.AddHs(mol)
# Generar coordenadas 3D si no están presentes
if not mol.GetConformer():
AllChem.EmbedMolecule(mol)
AllChem.UFFOptimizeMolecule(mol)
# Guardar como PDB
pdb_file = os.path.join(output_folder, f"{mol_name}.pdb")
Chem.MolToPDBFile(mol, pdb_file)
# Convertir a PDBQT usando Open Babel
subprocess.run(["obabel", pdb_file, "-O", pdbqt_file], check=True)
print(f"Generado: {pdbqt_file}")
# Eliminar el archivo PDB intermedio
os.remove(pdb_file)
print(f"Eliminado archivo intermedio: {pdb_file}")
# Ejecutar Vina para este archivo PDBQT
vina_output_file = os.path.join(vina_output_folder, f"5JJR_{mol_name}_complex.pdbqt")
# Imprimir el comando completo que se va a ejecutar
vina_command = [
vina_exe,
"--receptor", receptor_pdbqt,
"--ligand", pdbqt_file,
"--center_x", "27.523", "--center_y", "175.912", "--center_z", "18.436",
"--size_x", "15.581", "--size_y", "24.891", "--size_z", "12.990",
"--exhaustiveness", "32", "--out", vina_output_file,
"--num_modes", "10", "--spacing", "0.375", "--scoring", "vina", "--cpu", "8"
]
print(f"Ejecutando: {' '.join(vina_command)}")
# Ejecutar Vina y capturar su salida en tiempo real
try:
with open(deltaG_file, "a") as f, subprocess.Popen(vina_command, stdout=subprocess.PIPE, stderr=subprocess.PIPE, text=True) as process:
ligand_name = None
affinity_value = None
capturing_affinity = False # Controlar cuando empezar a capturar afinidad
# Leer línea por línea la salida de Vina
for line in process.stdout:
print(line, end="") # Imprimir en consola
# Buscar el ligando
if line.startswith("Ligand: "):
ligand_name = line.split()[1] # Obtener el nombre del ligando (después del espacio)
print(f"Encontrado ligando: {ligand_name}")
# Detectar la línea que indica el comienzo de los resultados
if line.startswith("-----+------------+----------+----------"):
capturing_affinity = True
print("Iniciando captura de afinidades...")
# Capturar la afinidad deltaG del primer modo
if capturing_affinity:
if line.strip(): # Ignorar líneas vacías
columns = line.split()
if len(columns) >= 2 and columns[0] == '1': # Verificar que es el primer modo
affinity_value = columns[1] # Obtener la afinidad (deltaG)
print(f"Encontrada afinidad: {affinity_value}")
if ligand_name and affinity_value:
# Guardar en deltaG.txt
f.write(f"{ligand_name}\t{affinity_value}\n")
print(f"Guardado en deltaG.txt: {ligand_name}\t{affinity_value}")
# Detener captura después del primer modo
capturing_affinity = False
# Esperar a que termine el proceso
process.wait()
print(f"Vina ejecutado para: {pdbqt_file}, resultado guardado en: {vina_output_file}")
except Exception as e:
print(f"Error ejecutando Vina para {pdbqt_file}: {e}")
# Registrar el archivo PDBQT procesado
with open(processed_file, "a") as f:
f.write(f"{pdbqt_file}\n")
# Eliminar el archivo PDBQT intermedio si lo deseas
# os.remove(pdbqt_file)
# print(f"Eliminado archivo intermedio: {pdbqt_file}")
# Rutas específicas para este caso
sdf_input_path = r"d:\vina-1.2.5\molecules.sdf" # Cambiar al nombre exacto del archivo SDF
output_directory = r"d:\vina-1.2.5\output_pdbqt"
vina_exe = r"d:/vina-1.2.5/vina_1.2.5_win.exe"
receptor_pdbqt = r"d:/vina-1.2.5/5JJR.pdbqt"
vina_output_directory = r"d:/vina-1.2.5/output"
deltaG_output_file = r"d:/vina-1.2.5/output_pdbqt/_deltaG.txt" # Archivo para guardar resultados de afinidad
# Llamar a la función
sdf_to_pdbqt_with_hydrogens(sdf_input_path, output_directory, vina_exe, receptor_pdbqt, vina_output_directory, deltaG_output_file)Versión para ejecutar en Linux.
# -*- coding: utf-8 -*-
import os
from rdkit import Chem
from rdkit.Chem import AllChem
import subprocess
def sdf_to_pdbqt_without_hydrogens(sdf_file, output_folder, vina_exe, receptor_pdbqt, vina_output_folder, deltaG_file):
"""
Convierte un archivo SDF a múltiples archivos PDBQT y ejecuta Vina para cada uno de los ficheros PDBQT generados.
Además, guarda un registro de los archivos procesados y extrae la afinidad (delta G) del archivo de salida de Vina.
conda install -c conda-forge vina
"""
# Crear la carpeta de salida si no existe
os.makedirs(output_folder, exist_ok=True)
os.makedirs(vina_output_folder, exist_ok=True)
# Leer moléculas del archivo SDF
supplier = Chem.SDMolSupplier(sdf_file)
# Crear o cargar el archivo de registro de archivos procesados
processed_file = os.path.join(output_folder, "processed_files.txt")
if os.path.exists(processed_file):
with open(processed_file, "r") as f:
processed_files = set(f.read().splitlines())
else:
processed_files = set()
# Verificar si podemos abrir el archivo deltaG.txt para escritura
try:
with open(deltaG_file, "a") as f:
pass
except Exception as e:
print(f"Error al intentar abrir el archivo {deltaG_file} para escritura: {e}")
return
for i, mol in enumerate(supplier):
if mol is None:
print(f"Molécula {i+1} no válida, saltando...")
continue
# Obtener el nombre de la molécula
mol_name = mol.GetProp("_Name") if mol.HasProp("_Name") else f"mol_{i+1}"
# Comprobar si el archivo PDBQT ya ha sido procesado
pdbqt_file = os.path.join(output_folder, f"{mol_name}.pdbqt")
if pdbqt_file in processed_files:
print(f"El archivo {pdbqt_file} ya ha sido procesado, saltando...")
continue
# Generar coordenadas 3D si no están presentes
if not mol.GetConformer():
AllChem.EmbedMolecule(mol)
AllChem.UFFOptimizeMolecule(mol)
# Guardar como PDB
pdb_file = os.path.join(output_folder, f"{mol_name}.pdb")
Chem.MolToPDBFile(mol, pdb_file)
# Convertir a PDBQT usando Open Babel
subprocess.run(["obabel", pdb_file, "-O", pdbqt_file], check=True)
print(f"Generado: {pdbqt_file}")
# Eliminar el archivo PDB intermedio
os.remove(pdb_file)
print(f"Eliminado archivo intermedio: {pdb_file}")
# Ejecutar Vina para este archivo PDBQT
vina_output_file = os.path.join(vina_output_folder, f"1P22_{mol_name}_complex.pdbqt")
vina_command = [
vina_exe,
"--receptor", receptor_pdbqt,
"--ligand", pdbqt_file,
"--center_x", "-1.549", "--center_y", "7.835", "--center_z", "9.745",
"--size_x", "23.813", "--size_y", "22.534", "--size_z", "27.981",
"--exhaustiveness", "32", "--out", vina_output_file,
"--num_modes", "10", "--spacing", "0.375", "--scoring", "vina", "--cpu", "8"
]
print(f"Ejecutando: {' '.join(vina_command)}")
try:
with open(deltaG_file, "a") as f, subprocess.Popen(vina_command, stdout=subprocess.PIPE, stderr=subprocess.PIPE, text=True) as process:
for line in process.stdout:
print(line, end="")
if line.startswith("Ligand: "):
ligand_name = line.split()[1]
if "-----+------------+----------+----------" in line:
columns = next(process.stdout).split()
if columns[0] == '1': # Primer modo
affinity_value = columns[1]
f.write(f"{ligand_name}\t{affinity_value}\n")
except Exception as e:
print(f"Error ejecutando Vina para {pdbqt_file}: {e}")
# Registrar el archivo PDBQT procesado
with open(processed_file, "a") as f:
f.write(f"{pdbqt_file}\n")
# Eliminar el archivo PDBQT intermedio
os.remove(pdbqt_file)
print(f"Eliminado archivo intermedio: {pdbqt_file}")
# Rutas específicas
sdf_input_path = "/home/jant/runDC/MP7-noQ-0117/MP7-noQ-0117.sdf"
output_directory = "/home/jant/runDC/MP7-noQ-0117/"
vina_exe = "/home/jant/vina-1.2.5/vina_1.2.5_linux_x86_64"
receptor_pdbqt = "/home/jant/runDC/MP7-noQ-0117/1P22-rec.pdbqt"
vina_output_directory = "/home/jant/runDC/MP7-noQ-0117/"
deltaG_output_file = "/home/jant/runDC/MP7-noQ-0117/_deltaG-MP7-noQ-0117.txt"
sdf_to_pdbqt_without_hydrogens(sdf_input_path, output_directory, vina_exe, receptor_pdbqt, vina_output_directory, deltaG_output_file)